|
To view this email as a web page, click here. |
 |
|
Welcome
Identifying unsuspected modifications in narrow window DIA data is possible with spectrum-centric searching.
In this month's highlighted publication, the authors investigate the spatial heterogeneity of uterine cancer tumors using laser microdissection.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
We have recently deleted a few predefined definitions of obsolete sequence databases.
Please have a read and feel free to contact us if you have any comments or questions.
|
|
|
|
|
|
 |
|
Mascot: The trusted reference standard for protein identification by mass spectrometry for 25 years
|
|
|
|
|
|
Finding unsuspected modifications in narrow window DIA data
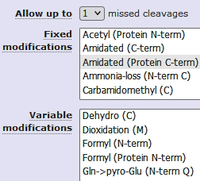
Most current DIA software is peptide-centric, requiring a spectral library for peptide identification. This often limits the maximum number of protein sequences, variable modifications and missed cleavages that can be accommodated. Mascot's spectrum-centric approach doesn't have these constraints. In particular, the Mascot error tolerant (ET) search is potentially a powerful approach to identify unsuspected sequence variants, modifications and non-specific enzyme cleavage products in narrow window DIA data.
We carried out an ET search on a PRIDE prostate cancer dataset, where the spectra were acquired in DIA mode with 8 m/z isolation window. The ET search is a two-stage process: the first pass search is performed using the search parameters specified in the search form, followed by a second pass with relaxed enzyme specificity, while iterating through a list of chemical and post-translational modifications and a residue substitution matrix.
The ET search found an additional 6835 significant PSMs and 1072 peptide sequences at 1% PSM FDR. These additional matches are the result of non-specific cleavages, deamidation, phosphorylation, and glycosylation as well as primary sequence variants.
Go to our blog to find out more.
|
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here, please send us a PDF or a URL.
|
|
|
Mapping three-dimensional intratumor proteomic heterogeneity in uterine serous carcinoma by multiregion microsampling
Allison L. Hunt, Nicholas W. Bateman, Waleed Barakat, Sasha C. Makohon-Moore, Tamara Abulez, Jordan A. Driscoll, Joshua P. Schaaf, Brian L. Hood, Kelly A. Conrads, Ming Zhou, Valerie Calvert, Mariaelena Pierobon, Jeremy Loffredo, Katlin N. Wilson, Tracy J. Litzi, Pang-Ning Teng, Julie Oliver, Dave Mitchell, Glenn Gist, Christine Rojas, Brian Blanton, Kathleen M. Darcy, Uma N. M. Rao, Emanuel F. Petricoin, Neil T. Phippen, G. Larry Maxwell & Thomas P. Conrads
Clinical Proteomics volume 21, Article number: 4 (2024)
The authors investigated the three-dimensional heterogeneity within the uterine serous carcinoma (USC) tumor microenvironment. They used spatially resolved laser microdissection (LMD) to select cellular subpopulations from nine patient tumor tissue specimens.
Specimen blocks were sectioned by cryotome into consecutive 10 µm tissue sections, and LMD was used to isolate enriched tumor epithelium (ET) or tumor-involved stroma (ES) cells in the 5 separate spatially distinct levels of the specimen block. LMD tissue underwent trypsin-digestion, and 5 µg of tryptic peptides per sample were labeled with isobaric 16-plex tandem mass tags. Samples were fractionated by basic reversed-phase LC prior to LC-MS/MS. Protein-level abundances were calculated from TMT reporter ion ratios from a minimum of two PSMs corresponding to a single protein accession.
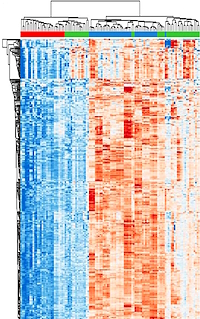
LC-MS/MS analysis quantified an average of 9548±450 proteins within each patient-specific TMT plex for a total of 6,503 proteins co-quantified across all nine patients. Unsupervised hierarchical cluster analysis of 351 variably abundant proteins (median absolute deviation >1) revealed two predominant branches with independent clustering of the ET samples from ES. Reverse phase protein microarray data showed 160 proteins that were co-quantified with MS.
They also compared the significantly altered proteins between ET and ES from the present USC study with those in their previous ovarian cancer dataset. They found a total of 313 proteins that were commonly altered and exhibited the same abundance trends between ET and ES.
|
|
|
|
|
|
|
|
|
Deleting obsolete database definitions
Mascot Server ships with predefined configuration for several sequence databases, stored in databases_1.xml. This includes bread-and-butter databases like SwissProt, but also some definitions that are now obsolete.
The International Protein Index (IPI) was compiled by the EBI back in 2004. The predefined definitions were added to databases_1.xml for Mascot Server 2.4, which is when the first version of Database Manager appeared. The IPI databases were superceded by Uniprot proteomes as long ago as September 2011, but we kept the definitions until now, because the FASTA files were still available. However, the FASTA files have now disappeared from the EBI website, so we have removed the IPI databases from databases_1.xml as well.
We have also removed the SARS-CoV-2 predefined definition. This was for a pre-release database that Uniprot created in 2020. The sequences have since then been reviewed and added to SwissProt, so this predefined definition has also been removed.
Removing the predefined definitions does not delete any databases in your local Mascot Server installation. If you had enabled any of the above databases, their local configuration points to the previous version of databases_1.xml and the sequence databases continue to work. The removal simply means you won’t be able to enable them on a new system or re-enable if you delete them. If you still need the databases, it’s best to make a local copy in Database Manager and make sure it’s part of your backups.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
|
|
|
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot. Read more about the company on our about page.
|




|
|
|
|
|
