|
To view this email as a web page, click here. |
|
|
|
Welcome
We're working on a universal spectrum-centric solution for the analysis of DIA data, called Mascot DIA.
This month's highlighted publication applies a metaproteomic workflow in a temperate marine ecosystem.
We are pleased to announce a price reduction to Mascot Server version updates.
|
|
|
|
|
|
 |
|
Mascot: The trusted reference standard for protein identification by mass spectrometry for 25 years
|
Get a quote
|
|
|
|
|
|
The promise of spectrum-centric DIA
|
|
|
We're working on a universal spectrum-centric solution for the analysis of DIA data, called Mascot DIA.
This solution is implemented using novel precursor detection in the upcoming Mascot Distiller 3.0, as well as new noise-resistant, probabilistic scoring in the upcoming Mascot Server 3.2.
Both products are currently in beta testing.
But what is spectrum-centric searching and what is it meant to solve?
"Spectrum-centric" and "peptide-centric" are complementary ways of analysing bottom-up LC-MS/MS data.
The goal of spectrum-centric analysis is to explain as many peaks in the MS/MS spectrum as possible.
If a statistically significant number of theoretical fragment peaks match the observed peaks, the precursor is considered identified.
In contrast, the goal of peptide-centric analysis is to find some evidence for a theoretical precursor in the LC-MS/MS run.
Most peptide-centric tools start from a library of theoretically detectable precursors, and they follow the elution of fragment peaks in the MS/MS scans.
If a sufficient number of highly correlated fragment traces are found, the precursor is considered identified.
The distinction can be subtle, but spectrum-centric searching has several benefits:
- Lower risk of false matches. A peptide identification demands a good run of b/y ions in the same spectrum, not just a handful of low-intensity fragments.
- All the information in the MS/MS spectrum is used, including neutral losses – essential for PTM localisation.
- With probabilistic scoring, there is no need to create a sample-specific spectral or chromatogram library.
- Because no library is needed, there is no restriction on variable modifications or choice of enzyme.
Head to
our blog
for a more detailed comparison.
|
|
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here, please send us a PDF or a URL.
|
|
|
Metaproteomic Dataset on Semi-Diurnal Variability of the Bacterioplankton Communities During a Spring Phytoplankton Bloom in the North Sea
Vaikhari Kale, Jürgen Bartel, Daniel Bartosik, Philip Berhard Lude, Chandni Sidhu, Hanno Teeling, Rudolf Amann, Thomas Schweder, Dörte Becher, Anke Trautwein-Schult
Proteomics, 0:e70001, 23 June 2025, doi:10.1002/pmic.70001
The authors use a metaproteomics workflow to study taxonomic and proteomic changes in the free-living bacterial community in phytoplankton blooms in the North Sea.
These can trigger bacterioplankton blooms, which play an important role in marine carbon cycling.
As just one example of the value of studying the diurnal cycle, compared to sampling once a day, the authors report significant changes in 65 relevant bacterial genera based on the summed protein group abundances between early and late samples.
They collected semi-diurnal samples across three days.
Seawater samples were filtered, and the bacterial community extracted, lysed and separated with SDS-PAGE.
After in-gel digestion, samples were analysed with LC-MS/MS in DDA mode.
A two-pass search strategy was used with Mascot Server.
The first pass results against a metagenome-derived database were combined and filtered in Scaffold, then a non-redundant subset database was created from matched proteins at the eight time points.
The second pass results against the combined subset database were filtered in Scaffold using stringent quality criteria.
Of the 20.5 million MS/MS spectra, 5.9 million were confidently identified with average identification rate of 28% per sample.
|

|
|
|
|
|
|
|
|
New pricing tiers for Mascot Server
|
|
|
We are pleased to announce the availability of Mascot Server under three new pricing tiers: Small (up to 8 cores), Medium (up to 16 cores) and Large (up to 128 cores).
The new tiers are better value for money with today's multicore processors than the old CPU-based licensing.
We have also reduced the cost to update from an older version.
If you have a licence for Mascot Server 2.x, no matter how old, you can trade it in for a 50% discount.
Also, the cost to update a 5 CPU or larger licence is significantly reduced when you switch to the Large tier, while drastically increasing the maximum core count.
If you have an active support contract, you may stay at your current CPU licence or switch to the new tier, whichever is better value for you.
We will contact all customers who are under support before their next support renewal date.
Please contact us if you would like more details or a formal quote.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot continues to be cited by over 2000 publications every year. Our software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Get a quote
|
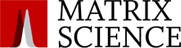
|
|
You can also contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|





|
|
|
|
|
