|
To view this email as a web page, click here. |
 |
|
Welcome
If you are going to the Human Proteome Organization World Congress in Taiwan next week, please stop by and visit us in booth 23.
We look at the reasons why the false discovery rate (FDR) for modified peptides can be much higher than for the search as a whole and what precautions you can take to minimise the problem.
This month's highlighted publication shows the deep characterization of a famous poxvirus.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month is an important update concerning the dropping of 'gi number' identifiers by NCBI.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
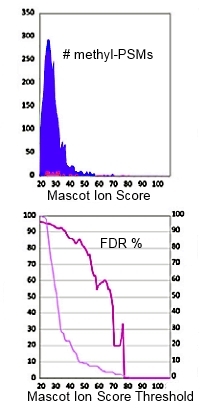
High FDR's for methylated peptides
Using a target/decoy search to estimate false discovery rate is an excellent validation method for MS/MS searches of large data sets, and has been extensively used by researchers for the past 10 years. However, a recent publication from the University of New South Wales reported FDR's for methylated peptides as high as 70%, even though the global FDR was less than 1%.
This important and well executed study raises some important questions concerning the accuracy and interpretation of database search results:
- We cannot assume that the global FDR applies to a subgroup of matches, such as modified peptides
- Target/decoy estimates the fraction of chance matches to unrelated sequences. It doesn’t model matches to homologous peptides or peptides with alternative arrangements of modifications
- We depend on competition to exclude certain types of false match
- Database search cannot tell you whether a modification has the correct elemental composition but the wrong structure or a modification is artefactual rather than post-translational
- FDRs can be based on counts of PSMs or counts of distinct sequences but don’t mix them; use one or the other consistently
- Combining sets of search results where the FDR is based on counts of distinct sequences is not straightforward
These factors are analysed over a series of three blog articles (part I, part II, part III). Some are limitations of the method that we can do little about. In other cases, there are ways to minimise the problem. In particular, deciding which modifications should be selected as fixed or variable and which can be left to be found by an error tolerant search. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
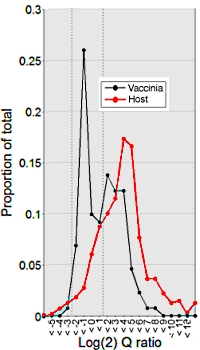
Protein primary structure of the Vaccinia virion at increased resolution
Tuan Ngo, Yeva Mirzakhanyan, Nissin Moussatche and Paul David Gershon
Journal of Virology, published online August 24, 2016
The vaccinia virus is a large, complex member of the poxvirus family, and was utilized in the eradication of smallpox. Though widely studied, the details of its protein components were not fully understood. The authors here undertook a very detailed analysis or the proteome of vaccinia with a variety of sample preparations and methods.
Four endoproeteases and CNBr digested the virion proteome, and LC-MS/MS with CID, HCD, or ETD followed by Mascot search produced new details on vaccinia. Additionally 18O-ATP labelling and IMAC preparation helped elucidate the phosphopeptides.
This resulted in the detection of over 88% of the theoretical proteome including the first-time detection of products from 27 open reading frames. Additionally the size of the characterized virion phosphoproteome was doubled from 189 to 396 unique phosphorylation sites. |
 |
|
|
|
|
|
|
|
Mascot tip of the month
NCBI have dropped 'gi number' identifiers from the 'nr' protein database a little earlier than previously announced.
The strengths of nr are that it is comprehensive and frequently updated. The downside is that it is a huge database. As of September 2016, the 54 GB Fasta file contained 94 million entries. In most cases, there are better choices of database, such as a subset of GenBank for the organism of interest or a Uniprot complete proteome.
If you are part way through a major project or have a workflow that absolutely requires the continued use of gi numbers as identifiers, you will need to freeze nr at or before the 21 August 2016 release. That is, you must disable any type of automatic updating.
The nr Fasta and taxonomy index files currently available for download from NCBI contain some errors and omissions. We have posted new configuration details, but it is likely that we will be able to improve on these in a few weeks time, as things settle down, so don't rush to update this database on your in-house Mascot Server. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
