|
To view this email as a web page, click here. |
 |
|
Welcome
Learn about the features of Mascot Server and Mascot Distiller that enable successful identifications to be obtained from chimeric spectra.
This month's highlighted publication shows a high resolution structure of the yeast ribosome-assembly machinery.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month explains the special treatment given to initiator methionine.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
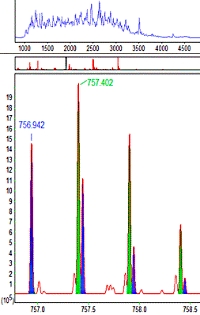
Untangling chimeric spectra
If two peptides with different sequences have very similar masses and retention times, they may be co-fragmented to give a so-called chimeric MS/MS spectrum, containing sequence ions from both. In most cases, modern high resolution instruments will be able to resolve the fragments even though the isotopic distributions are overlapped. If so, Mascot Distiller can determine the individual peptide masses and output them to the peak list
Mascot Server will use all the peptide masses when searching the MS/MS data. If it gets significant matches to more than one sequence, these are reported independently. If there is just one match, it is associated with the closest peptide mass.
To evaluate this approach, a publicly available yeast dataset was peak picked by Mascot Distiller, allowing for up to 4 precursors per MS/MS spectrum, searched, and the results reported at 1% peptide FDR. A total of 342,827 MS/MS spectra gave 207,882 significant peptide matches. This compares with 167,806 matches when only a single precursor was allowed, an increase of over 20%.
Please go here to read more about how you can take advantage of this ability to handle multiple precursors. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
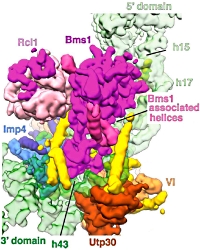
Architecture of the yeast small subunit processome
Malik Chaker-Margot, Jonas Barandun, Mirjam Hunziker, Sebastian Klinge
Science 13 Jan 2017: Vol. 355, Issue 6321
The small subunit (SSU) processome is a large ribonucleoprotein complex composed of approximately 70 non-ribosomal proteins, several small nucleolar RNA's, and ribosomal RNA precursors (pre-rRNA). It organizes the assembly of the eukaryotic small ribosomal subunit at the early stages by coordinating the folding and modification of pre-rRNA.
The authors used cryo-electron microscopy to elucidate the structure of the S. cerevisiae SSU processome at a resolution of 5.1 Å. They were able to show how the molecular scaffold provided by the 5' external transcribed spacers (ETS) and U3 small nucleoloar RNA is used as a foundation for the assembly of the small eukaryotic ribosomal subunit.
They were also able to show that proteins that bind within the 5' ETS are located at the base of the SSU processome structure, those associated with 18S rRNA domains form the core of the particle, and proteins recruited only after 18S rRNA completion form the outer tier of the structure.
|
 |
|
|
|
|
|
|
|
Mascot tip of the month
When a protein is translated from mRNA, the start codon in most genetic codes corresponds to methionine. Virtually all sequences in protein databases are machine translations of experimentally determined nucleic acid sequences, so this applies to most of the protein sequences that we search. In the Jan 2017 SwissProt, 97.5% of the 553,474 entries started with M.
Mature proteins are often modified, post-translationally, by a methionine aminopeptidase, to remove the N-terminal Met. Because this is so common, Mascot Server has this possibility hard coded. That is, if the database sequence begins with M, whatever the enzyme specified for the search, Mascot will test the N-terminal peptide with and without the initiator Met. Note that the mature protein amino terminus may be further modified by the addition of acetyl, propionyl or methyl groups, or membrane anchors, such as palmitoyl and myristoyl. In most cases, if this is of interest, you will want to test for these possibilities using an error tolerant search, rather than burden the entire search with a large number of variable modifications.
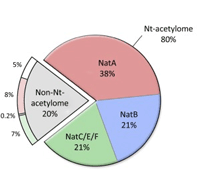
A further advantage of using an error tolerant search is that it can pick up cases where the mature protein is created by removal of an extended signal peptide, because it relaxes the selected enzyme to be semi-specific. Since an error tolerant search does not allow a peptide to be both semi-specific and carry an unsuspected modification, best to include Acetyl (Protein N-term) as a variable modification, because it has been estimated that 80% of all human proteins are N-term acetylated. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
