|
To view this email as a web page, click here. |
 |
|
Welcome
We discuss how spectral library searching can play an important role in your current workflows.
This month's highlighted publication shows a new approach for localizing proteins in organelles.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month describes how to create and modify enzyme definitions for Mascot Server.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Use spectral libraries today
A recent report on the use of spectral libraries in proteomics identified challenges that could be hindering wider adoption of the method. We suggest that these supposed barriers simply aren't an issue when using Mascot Server.
- Integrating library search into existing workflows is difficult With Mascot Server, spectral library searching is integrated seamlessly from the search form through to the results report and data export. If you need to write your own code to read Mascot result files, Mascot Parser enables library search results to be handled in exactly the same way as Fasta search results.
- File formats are still evolving Mascot Server integrates NIST’s well-established MSPepSearch, which supports the MSP file format. Each spectrum is a standalone entry and contains, at minimum, a peak list and a few key-value fields describing the precursor mass and peptide sequence. The MSP format is flexible and perfectly adequate for current needs.
- No comprehensive community libraries The report presents a vision for a community library that contains 'everything'. Importing and curation of results is fully automated, the pedigree of each spectrum can be traced precisely and each spectrum represents the ultimate synthesis for how the peptide fragments. This has more in common with a repository than with a library for day-to-day searching. There are many libraries available for download. If none of these match your experiment exactly - maybe you are studying an unusual organism or using a quantitation tag - Mascot Server makes it relatively easy to create a library from your own search results.
To read more about how you can use Mascot Server for library searching today, click here.
|
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
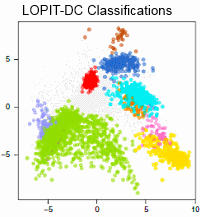
Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics
Aikaterini Geladaki, Nina Kocevar Britovsek, Lisa M. Breckels, Tom S. Smith, Owen L. Vennard, Claire M. Mulvey, Oliver M. Crook, Laurent Gatto & Kathryn S. Lilley
Nature Communications, volume 10, Article number: 331 (2019)
This paper introduces a method for high-confidence localization of proteins in suborganellar structures, protein complexes, and signaling pathways. Localization of Organelle Proteins by Isotope Tagging after Differential ultraCentrifugation (LOPIT-DC) is a spatial proteomics pipeline based on differential centrifugation, as opposed to the density gradient-based hyperLOPIT approach.
The workflow entails cell fractionation, organelle separation into 10 fractions by differential centrifugation, peptide generation and TMT labelling, followed by LC-MS/MS and MS3 for quantitation. LOPIT-DC makes use of TMT multiplexing, simultaneously analyzing all subcellular fractions in a single MS run.
The authors compare protein subcellular localization maps produced by LOPIT-DC and hyperLOPIT using the human osteosarcoma U-2 OS cell line. LOPIT-DC achieved high subcellular resolution and comparable classifications to hyperLOPIT using less sample, time and resources. Where resources are not limited, hyperLOPIT provides the maximum possible overall subcellular resolution but in cases of starting material, time or financial constraints, the simpler and quicker LOPIT-DC protocol is a good alternative.
|
 |
|
|
|
|
|
|
|
Mascot Tip
You can easily modify existing enzyme definitions or create new ones in the Mascot Server configuration editor. Starting from your local Mascot Server home page, choose Configuration Editor then Enzymes.
It's unlikely that you'll need to add a brand new protease, but if you are using multiple enzymes, you may need to create a custom enzyme mixture. For example, imagine you are using CNBr (cuts C-term to Met) followed by Asp-N (cuts N-term to D).
Choose Add new enzyme and give the new mixture a suitable title, e.g. CNBr+Asp-N. Under Components, add a second row and enter the two specificities. For Asp-N, you can might choose to cleave at D alone, but it is safer to enter BD, because B is the ambiguity code for D or N.
There are two important checkboxes towards the top of the form. If Independent is not checked, the protein is cleaved as if multiple enzymes had been added to a single sample aliquot. This means that one end of a peptide could correspond to one cutter and the other end to a different cutter. If Independent is checked, this simulates independent digests on separate sample aliquots, which are later mixed, so that individual peptides correspond to just one of the cutters. You can see the difference by using the Test Enzyme button.
If Semispecific is checked, peptides are allowed to be non-specific at one terminus. This creates a much larger search space, and should only be used when essential. For this example, if Independent is unchecked, the number of peptides expected from the default test protein increases from 9 to 391 when Semispecific is checked. Unless you anticipate a very high level of non-specific cleavage, better to use an error tolerant search, which relaxes the enzyme specificity in the second pass.
As with other modules in the Configuration Editor, help is available for most fields by hovering the mouse cursor over the control or label.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
