|
To view this email as a web page, click here. |
 |
|
Welcome
Mascot Server 2.7 is now released, and those of you under warranty or support have been sent a free update key and download link by email. If you believe you should have received an update and haven't, first check your spam folder. If still no joy, please contact us.
Long-term reproducibility of proteomics workflows can be a problem, and we have some suggestions to help you.
This month's highlighted publication shows that the addition of ion mobility can improve sequence coverage of histone proteoforms.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month describes how Mascot Server handles non-standard modification names in a spectral library search.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Is your database search reproducible?
Instrument and process variability are usually the main factors underlying poor reproductibility in proteomics workflows, but data analysis is also critical. In particular, it is important to record all the software parameters used, and ensure the software produces the same output given the same input and parameters.
To facilitate reproducible data analysis, Mascot Server automatically records the complete set of metadata describing the search conditions to the results file. This encompasses search parameters, processing options, filepaths, versions, definitions, rules, quant methods, as well as the input peak lists. The one thing the results file does not store is the protein Fasta. Doing so would greatly inflate its size and create serious storage issues.
Finally, you need to have a high level of compatibility between software versions. It is important that updating to a new version and repeating a search should not have a major impact on the results. Sometimes, things will change because of a bug fix, but when a new feature is introduced, we try to include a mechanism to override or disable the new behaviour.
For details on the factors affecting reproducibility in your lab's work, go here to read more.
|
|
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Middle-Down Proteomic Analyses with Ion Mobility Separations of Endogenous Isomeric Proteoforms
Pavel V. Shliaha, Vladimir Gorshkov, Sergey I. Kovalchuk, Veit Schwammle, Matthew A. Baird, Alexandre A. Shvartsburg, and Ole N. Jensen
Analytical Chemistry 92 2364-8 (2020)
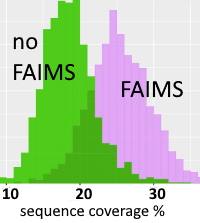
The authors have investigated the use of field asymmetric waveform ion mobility spectrometry (FAIMS) to augment the separation of the complex sets of proteoforms present in histones. The diversity of structures and isomers results from the presence of numerous methylations, acetylations, phosphorylations, and other PTMs on alternative sites. Conventional LC separations are often insufficient to separate proteoforms that only differ in the location of PTMs.
Histones were extracted from mouse embryonic stem cells and the H3 family was purified by LC. Histone tails were severed by Glu-C and analyzed by LC-[FAIMS]MS/MS using ETD, with identification by Mascot search against the MS_HistoneDB database. The LC runs were repeated for 9 increasing voltages of the FAIMS device and the whole analysis (including the column equilibration) took approx. 30 hours. The authors discovered 526 N-tail proteoforms in total, in comparison to some 200 forms for LC-MS/MS acquisitions with no FAIMS.
|
 |
|
|
|
|
|
|
|
Mascot Tip
Some spectral library files contain non-standard modification names, meaning the Mascot peptide view report cannot show a table of fragment masses or label the spectrum. In Mascot Server 2.7, we have introduced a new configuration file called library_mod_aliases that contains mappings between names found in spectral library files and Unimod modification names.
The syntax is name_used_in_library_file = Unimod_name. Any modification in any spectral library with the name shown on the left will be replaced with the name on the right before being saved to the .nist_format file, used by MSPepSearch. There isn't a browser-based editor for library_mod_aliases, as yet; you have to use a text editor.
On installation, the file is populated with some common examples found in PRIDE and NIST libraries, but it is quite likely that you will have to add new entries if you do much library searching. PRIDE is increasingly using mass values to annotate entries, rather than modification names. This is a retrograde step, since rounding errors lead to spurious modifications. In PRIDE_Contaminants, for example, Nethylmaleimide is sometimes represented as 125.047678 and sometimes as 125.047679.
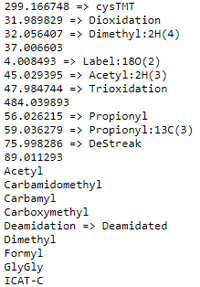
You can see which names are recognised and which are not by following the statistics link from the Database Status page. An excerpt from the statistics file for PRIDE_Contaminants is shown on the right. Items with an arrow have been mapped to Unimod names, while those without have not. If these are non-Unimod names, such as 37.006603, there will be no fragment mass information in peptide view.
|
|
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
