
Proteomics of Primary Uveal Melanoma: Insights into Metastasis and Protein Biomarkers

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Primary UM Tumor Samples
2.2. Proteomic Characterization of the Choroid Controls
2.3. pUM Quantitative Proteomics Overview
2.4. Independent Evidence Supporting the iTRAQ Protein Quantitation
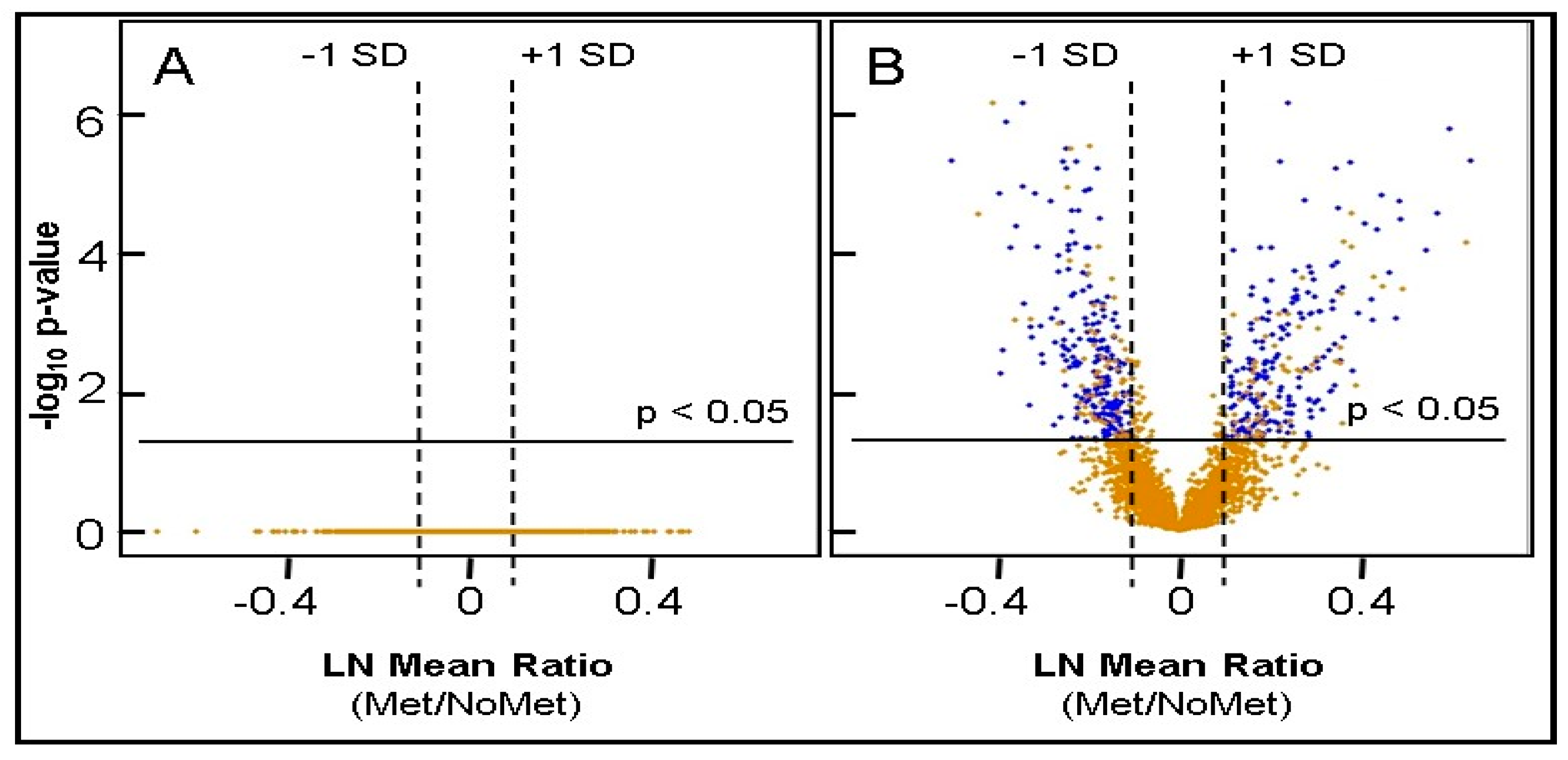
2.5. Identification of Differentially Expressed Proteins
2.6. Bioinformatic Differences between Metastasizing and Non-Metastasizing pUM
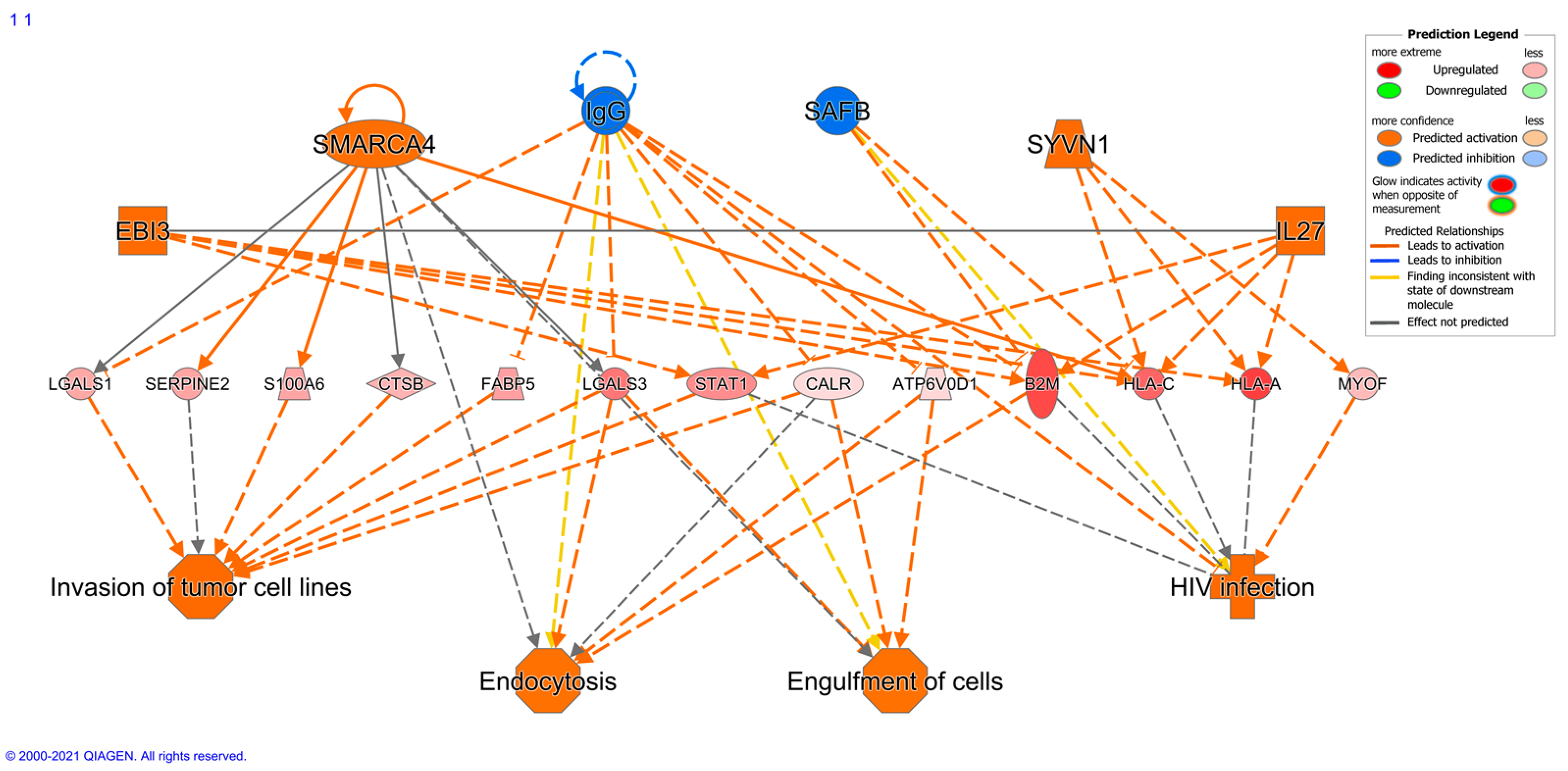
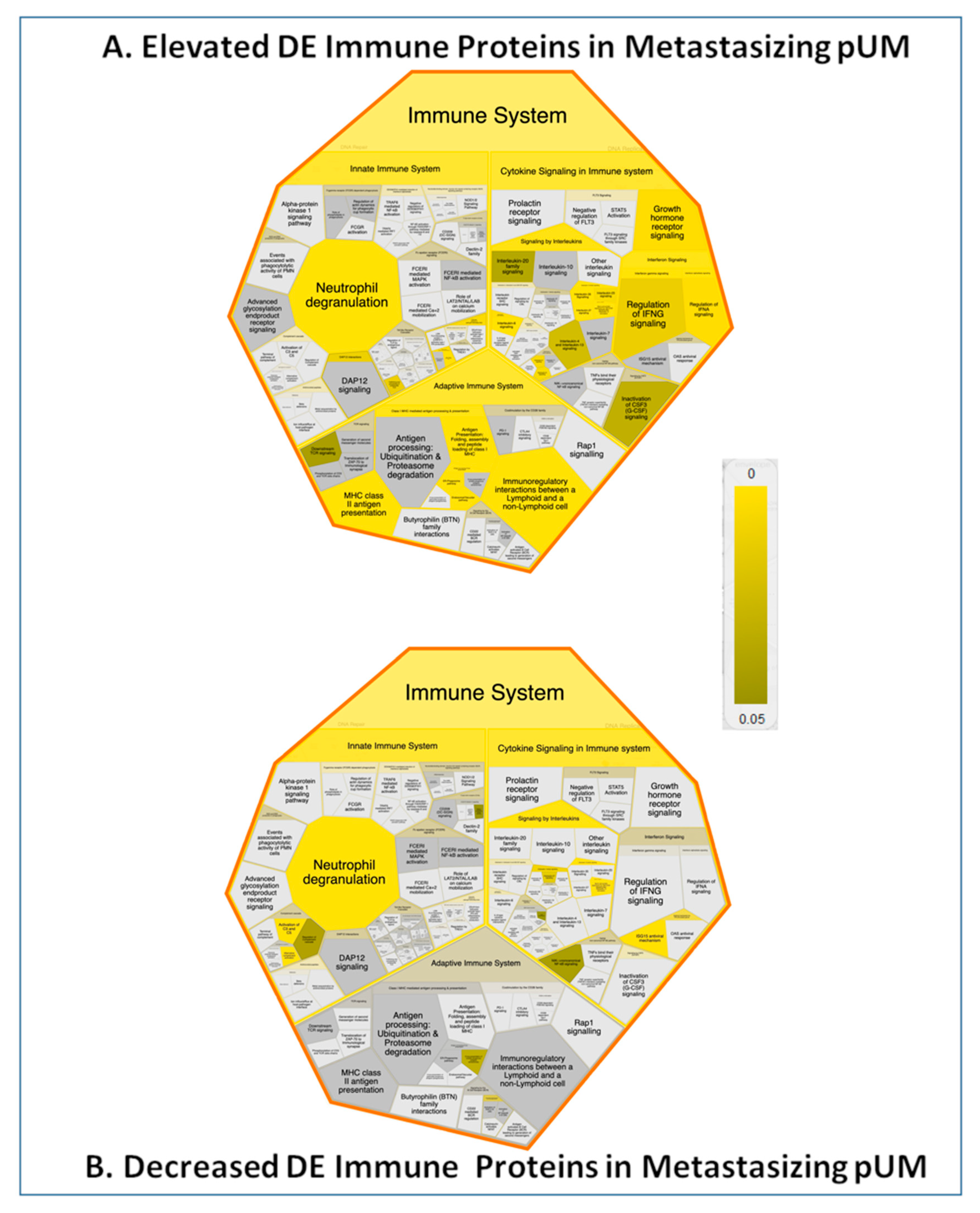
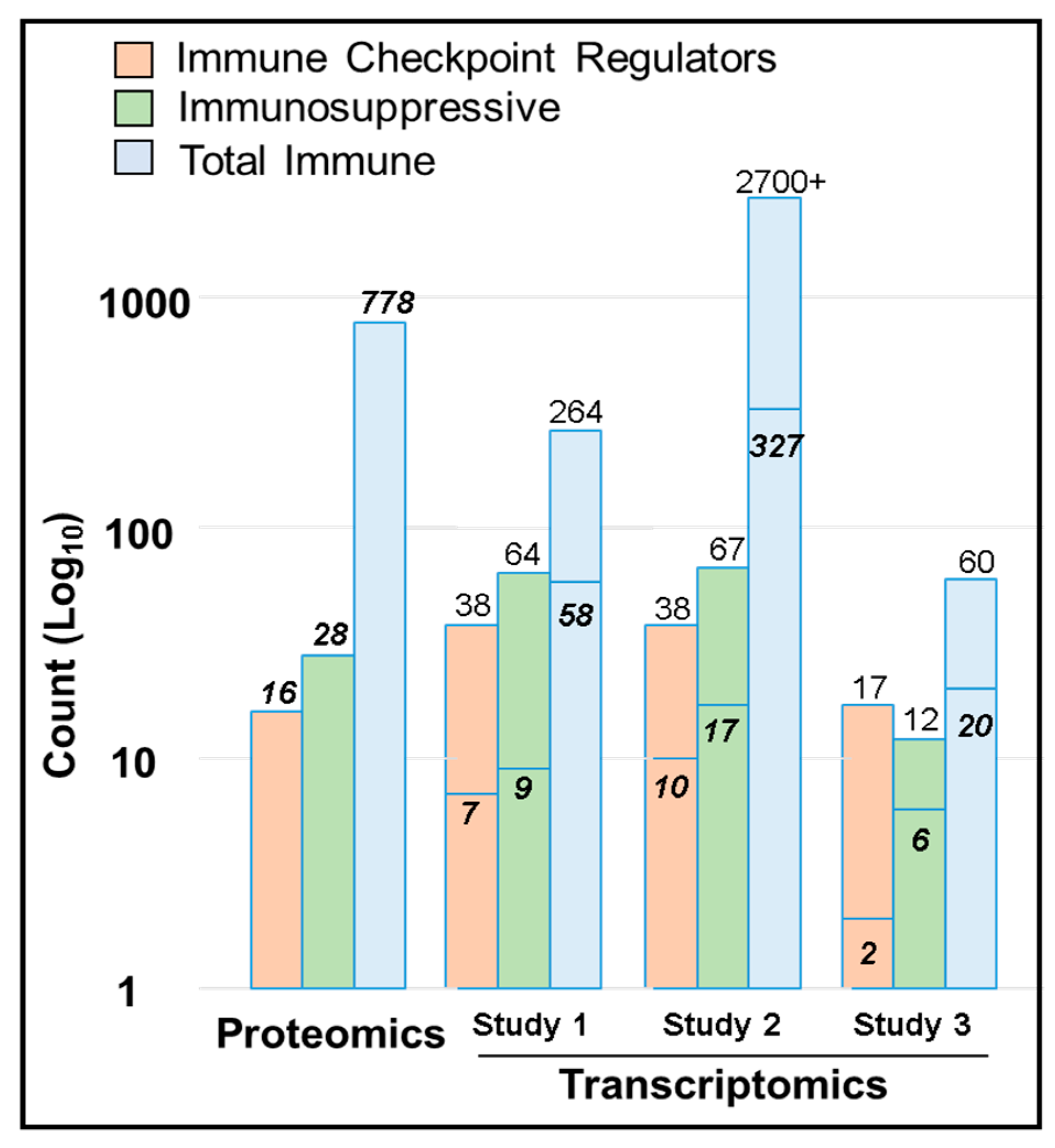
2.7. Immune Protein Profiling
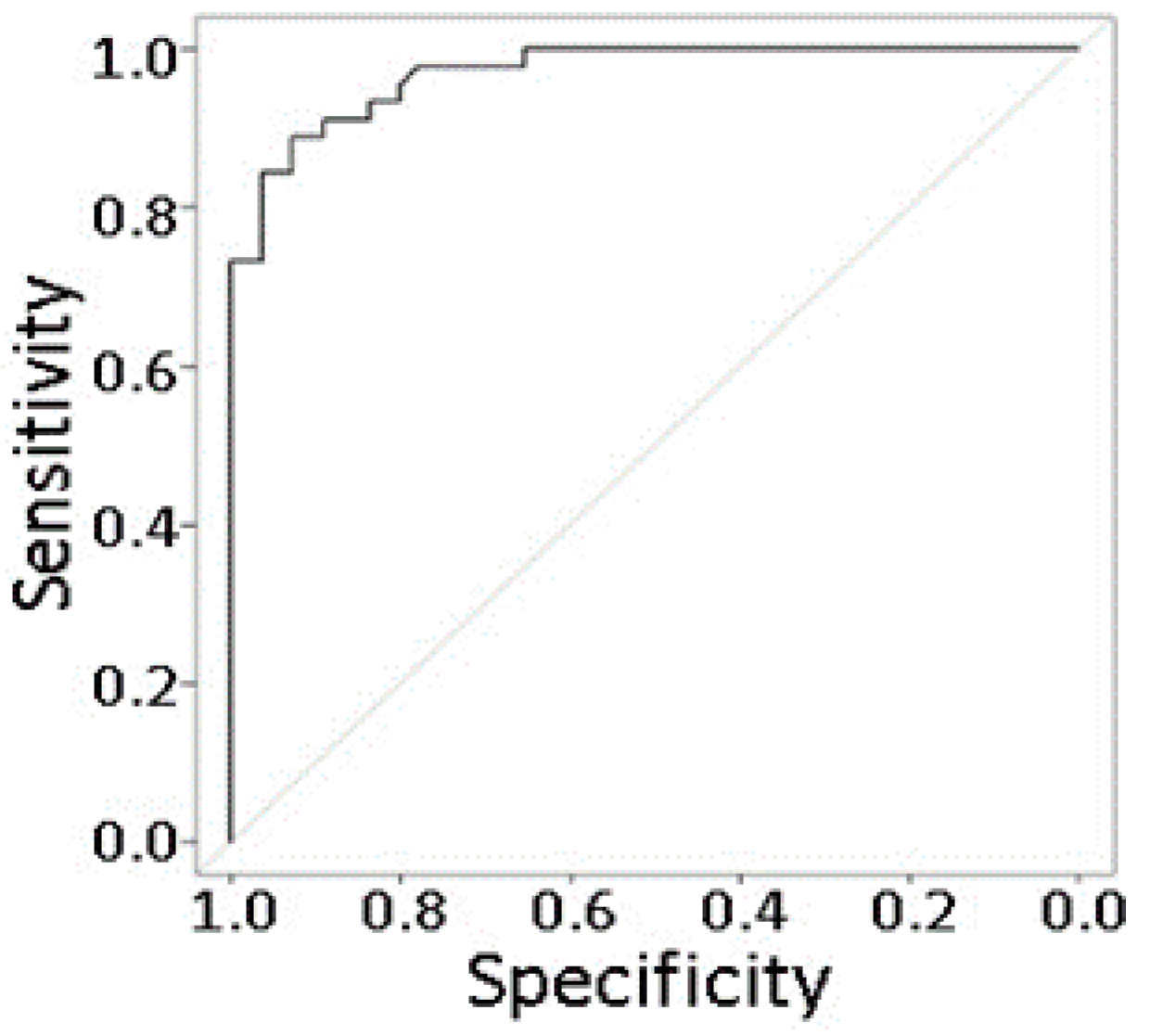
2.8. Prediction Modeling
3. Discussion
4. Materials and Methods
4.1. Specimens
4.2. Sample Preparation
4.3. ITRAQ Labeling and Peptide Fractionation
4.4. Protein Identification
4.5. Protein Quantitation
4.6. Statistical Analysis
4.7. Bioinformatics
4.8. Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thorton, S.; Kalirai, H.; Aughton, K.; Coupland, S.E. Unpacking the genetic etiology of uveal melanoma. Expert Rev. Ophthalmol. 2020, 15, 211–220. [Google Scholar] [CrossRef]
- Collaborative Ocular Melanoma Study Group. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch. Ophthalmol. 2006, 124, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W. The genetics of uveal melanoma: An emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012, 25, 171–181. [Google Scholar] [CrossRef]
- Kath, R.; Hayungs, J.; Bornfeld, N.; Sauerwein, W.; Hoffken, K.; Seeber, S. Prognosis and treatment of disseminated uveal melanoma. Cancer 1993, 72, 2219–2223. [Google Scholar] [CrossRef]
- Collaborative Ocular Melanoma Study Group. The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma I: Characteristics of patients enrolled and not enrolled. COMS report no. 9. Am. J. Ophthalmol. 1998, 125, 767–778. [Google Scholar] [CrossRef]
- Harbour, J.W. Molecular prognostic testing and individualized patient care in uveal melanoma. Am. J. Ophthalmol. 2009, 148, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalirai, H.; Coupland, S.E. An Update on Ocular Melanoma. Diagn. Histopathol. 2014, 21, 19–25. [Google Scholar] [CrossRef]
- Van den Bosch, T.; Kilic, E.; Paridaens, D.; de Klein, A. Genetics of uveal melanoma and cutaneous melanoma: Two of a kind? Dermatol. Res. Pract. 2010, 2010, 360136. [Google Scholar] [CrossRef] [Green Version]
- Komatsubara, K.M.; Carvajal, R.D. Immunotherapy for the Treatment of Uveal Melanoma: Current Status and Emerging Therapies. Curr. Oncol. Rep. 2017, 19, 45. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Wilson, D.; Finger, P.T.; Worley, L.A.; Onken, M.D. Gene expressing profiling of iris melanomas. Ophthalmology 2013, 120, 213.e3. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, P.; Murphy, C.C.; Clynes, M.; Horgan, N.; Moriarty, P.; Tiernan, D.; Beatty, S.; Kennedy, S.; Meleady, P. Proteomics in uveal melanoma. Exp. Eye Res. 2014, 118, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.; Duke, C.; Coupland, S.E.; Hiscott, P.; Smith, P.A.; Campbell, I.; Douglas, A.; Howard, P. Cytogenetics of uveal melanoma: A 7-year clinical experience. Ophthalmology 2007, 114, 1925–1931. [Google Scholar] [CrossRef]
- Herlihy, N.; Dogrusoz, M.; van Essen, T.H.; Harbour, J.W.; van der Velden, P.A.; van Eggermond, M.C.; Haasnoot, G.W.; van den Elsen, P.J.; Jager, M.J. Skewed expression of the genes encoding epigenetic modifiers in high-risk uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1447–1458. [Google Scholar] [CrossRef] [Green Version]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, C.R.; Kalirai, H.; Sacco, J.J.; Azzevedo, R.A.; Duckworth, A.; Slupsky, J.R.; Coulson, J.M.; Coopland, S.E. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J. Pathol. 2020, 250, 420–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, Y.; Acha-Sagredo, A.; Sabat-Pospiech, D.; Kipling, N.; Clarke, K.; Figueiredo, C.R.; Kalirai, H.; Coupland, S.E. Transcriptome Profiling Reveals New Insights into the Immune Microenvironment and Upregulation of Novel Biomarkers in Metastatic Uveal Melanoma. Cancers 2020, 12, 2832. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Person, E.; Char, D.H.; Bowcock, A.M.; Harbour, J.W. Loss of Heterozygosity of Chromosome 3 Detected with Single Nucleotide Polymorphisms Is Superior to Monosomy 3 for Predicting Metastasis in Uveal Melanoma. Clin. Cancer Res. 2007, 13, 2923–2927. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.D.; Aronow, M.E.; Sun, Y.; Bebek, G.; Saunthararajah, Y.; Schoenfield, L.R.; Biscotti, C.V.; Tubbs, R.R.; Triozzi, P.L.; Eng, C. Chromosome 3 status in uveal melanoma: A comparison of fluorescence in situ hybridization and single-nucleotide polymorphism array. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3331–3339. [Google Scholar] [CrossRef] [Green Version]
- Damato, B.; Dopierala, J.; Klaasen, A.; van Dijk, M.; Sibbring, J.; Coupland, S.E. Multiplex ligation-dependent probe amplification of uveal melanoma: Correlation with metastatic death. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3048–3055. [Google Scholar] [CrossRef] [Green Version]
- Onken, M.D.; Worley, L.A.; Tuscan, M.D.; Harbour, J.W. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J. Mol. Diagn. 2010, 12, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Char, D.H.; Augsburger, J.J.; Correa, Z.M.; Nudleman, E.; Aaberg, T.M., Jr.; Altaweel, M.M.; Bardenstein, D.S.; Finger, P.T.; et al. Collaborative Ocular Oncology Group report number 1: Prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology 2012, 119, 1596–1603. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W. A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol. Biol. 2014, 1102, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef]
- Thornton, S.; Coupland, S.E.; Olohan, L.; Sibbring, J.S.; Kenny, J.G.; Hertz-Fowler, C.; Liu, X.; Haldenby, S.; Heimann, H.; Hussain, R.; et al. Targeted Next-Generation Sequencing of 117 Routine Clinical Samples Provides Further Insights into the Molecular Landscape of Uveal Melanoma. Cancers 2020, 12, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogrusoz, M.; Jager, M.J. Genetic prognostication in uveal melanoma. Acta Ophthalmol. 2018, 96, 331–347. [Google Scholar] [CrossRef]
- Martel, A.; Baillif, S.; Nahon-Esteve, S.; Gastaud, L.; Bertolotto, C.; Romeo, B.; Mograbi, B.; Lassalle, S.; Hofman, P. Liquid Biopsy for Solid Ophthalmic Malignancies: An Updated Review and Perspectives. Cancers 2020, 12, 3284. [Google Scholar] [CrossRef]
- Crabb, J.W.; Hu, B.; Crabb, J.S.; Triozzi, P.; Saunthararajah, Y.; Tubbs, R.; Singh, A.D. iTRAQ Quantitative Proteomic Comparison of Metastatic and Non-Metastatic Uveal Melanoma Tumors. PLoS ONE 2015, 10, e0135543. [Google Scholar] [CrossRef] [Green Version]
- Coupland, S.E.; Vorum, H.; Mandal, N.; Kalirai, H.; Honore, B.; Urbak, S.F.; Lake, S.L.; Dopierala, J.; Damato, B.E. Proteomics of uveal melanomas suggests HSP-27 as a possible surrogate marker of chromosome 3 loss. Investig. Ophthalmol. Vis. Sci. 2010, 51, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linge, A.; Kennedy, S.; O’Flynn, D.; Beatty, S.; Moriarty, P.; Henry, M.; Clynes, M.; Larkin, A.; Meleady, P. Differential expression of fourteen proteins between uveal melanoma from patients who subsequently developed distant metastases versus those who did Not. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4634–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missotten, G.S.; Beijnen, J.H.; Keunen, J.E.; Bonfrer, J.M. Proteomics in uveal melanoma. Melanoma Res. 2003, 13, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Garcia, A.; Thomas, B.; Pineiro, A.; Akoulitchev, A.; Dwek, R.A.; Zitzmann, N. Proteome analysis of a human uveal melanoma primary cell culture by 2-DE and MS. Proteomics 2005, 5, 4980–4993. [Google Scholar] [CrossRef]
- Pardo, M.; García, Á.; Thomas, B.; Piñeiro, A.; Akoulitchev, A.; Dwek, R.A.; Zitzmann, N. The characterization of the invasion phenotype of uveal melanoma tumour cells shows the presence of MUC18 and HMG-1 metastasis markers and leads to the identification of DJ-1 as a potential serum biomarker. Int. J. Cancer 2006, 119, 1014–1022. [Google Scholar] [CrossRef]
- Pardo, M.; García, A.; Antrobus, R.; Blanco, M.; Dwek, R.; Zitzmann, N. Biomarker discovery from uveal melanoma secretomes: Identification of gp100 and cathepsin D in patient serum. J. Proteome Res. 2007, 6, 2802–2811. [Google Scholar] [CrossRef]
- Zuidervaart, W.; Hensbergen, P.J.; Wong, M.C.; Deelder, A.M.; Tensen, C.P.; Jager, M.J.; Gruis, N.A. Proteomic analysis of uveal melanoma reveals novel potential markers involved in tumor progression. Investig. Ophthalmol. Vis. Sci. 2006, 47, 786–793. [Google Scholar] [CrossRef]
- Wang, F.; Bing, Z.; Zhang, Y.; Ao, B.; Zhang, S.; Ye, C.; He, J.; Ding, N.; Ye, W.; Xiong, J.; et al. Quantitative proteomic analysis for radiation-induced cell cycle suspension in 92-1 melanoma cell line. J. Radiat. Res. 2013, 54, 649–662. [Google Scholar] [CrossRef]
- Yan, L.B.; Shi, K.; Bing, Z.T.; Sun, Y.L.; Shen, Y. Proteomic analysis of energy metabolism and signal transduction in irradiated melanoma cells. Int. J. Ophthalmol. 2013, 6, 286–294. [Google Scholar] [CrossRef]
- Angi, M.; Kalirai, H.; Prendergast, S.; Simpson, D.; Hammond, D.E.; Madigan, M.C.; Beynon, R.J.; Coupland, S.E. In-depth proteomic profiling of the uveal melanoma secretome. Oncotarget 2016, 7, 49623–49635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsering, T.; Laskaris, A.; Abdouh, M.; Bustamante, P.; Parent, S.; Jin, E.; Ferrier, S.T.; Arena, G.; Burnier, J.V. Uveal Melanoma-Derived Extracellular Vesicles Display Transforming Potential and Carry Protein Cargo Involved in Metastatic Niche Preparation. Cancers 2020, 12, 2923. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, Y.; Zou, L.; Wang, D.; Shao, C.; Wang, Y.; Sun, W.; Zhang, L. A Proteomic Analysis of Individual and Gender Variations in Normal Human Urine and Cerebrospinal Fluid Using iTRAQ Quantification. PLoS ONE 2015, 10, e0133270. [Google Scholar] [CrossRef] [Green Version]
- Higdon, R.; Kolker, E. Can “normal” protein expression ranges be estimated with high-throughput proteomics? J. Proteome Res. 2015, 14, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Radivoyevitch, T.; Zabor, E.C.; Singh, A.D. Uveal Melanoma: Long-Term Survival. PLoS ONE 2021. [Google Scholar] [CrossRef]
- Singh, A.D.; Zabor, E.C.; Radivoyevitch, T. Estimating Cured Fractions of Uveal Melanoma. JAMA Ophthalmol. 2021, 139, 174–181. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Valet, F.; Planque, N.; Silveri, L.; Maacha, S.; Anezo, O.; Hupe, P.; Plancher, C.; Reyes, C.; Albaud, B.; et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011, 71, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Tian, Q.; Stepaniants, S.B.; Mao, M.; Weng, L.; Feetham, M.C.; Doyle, M.J.; Yi, E.C.; Dai, H.; Thorsson, V.; Eng, J.; et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol. Cell. Proteom. 2004, 3, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Jmor, F.; Kalirai, H.; Taktak, A.; Damato, B.; Coupland, S.E. HSP-27 protein expression in uveal melanoma: Correlation with predicted survival. Acta Ophthalmol. 2012, 90, 534–539. [Google Scholar] [CrossRef]
- De Waard-Siebinga, I.; Kool, J.; Jager, M.J. HLA antigen expression on uveal melanoma cells in vivo and in vitro. Hum. Immunol. 1995, 44, 111–117. [Google Scholar] [CrossRef]
- Gangemi, R.; Mirisola, V.; Barisione, G.; Fabbi, M.; Brizzolara, A.; Lanza, F.; Mosci, C.; Salvi, S.; Gualco, M.; Truini, M.; et al. Mda-9/syntenin is expressed in uveal melanoma and correlates with metastatic progression. PLoS ONE 2012, 7, e29989. [Google Scholar] [CrossRef]
- Jha, J.; Singh, M.K.; Singh, L.; Pushker, N.; Bajaj, M.S.; Sen, S.; Kashyap, S. Prognostic relevance of ATM protein in uveal melanoma and its association with clinicopathological factors. Int. J. Clin. Oncol. 2019, 24, 1526–1535. [Google Scholar] [CrossRef]
- Narasimhaiah, D.; Legrand, C.; Damotte, D.; Remark, R.; Munda, M.; De Potter, P.; Coulie, P.G.; Vikkula, M.; Godfraind, C. DNA alteration-based classification of uveal melanoma gives better prognostic stratification than immune infiltration, which has a neutral effect in high-risk group. Cancer Med. 2019, 8, 3036–3046. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, C.; Oba, J.; Roszik, J.; Marszalek, J.R.; Chen, K.; Qi, Y.; Eterovic, K.; Robertson, A.G.; Burks, J.K.; McCannel, T.A.; et al. Elevated Endogenous SDHA Drives Pathological Metabolism in Highly Metastatic Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4187–4195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Merbs, S.L.; Sokoll, L.J.; Chan, D.W.; Zhang, Z. A multiplex immunoassay of serum biomarkers for the detection of uveal melanoma. Clin. Proteom. 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez, G.; Nguyen, H.V.; Chemudupati, T.; Ludwig, C.A.; Toral, M.; Reddy, S.; Mruthyunjaya, P.; Mahajan, V.B. Liquid biopsy proteomics of uveal melanoma reveals biomarkers associated with metastatic risk. Mol. Cancer 2021, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Achberger, S.; Aldrich, W.; Tubbs, R.; Crabb, J.W.; Singh, A.D.; Triozzi, P.L. Circulating immune cell and microRNA in patients with uveal melanoma developing metastatic disease. Mol. Immunol. 2014, 58, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Triozzi, P.L.; Aldrich, W.; Crabb, J.W.; Singh, A.D. Spontaneous cellular and humoral tumor antigen responses in patients with uveal melanoma. Melanoma Res. 2015, 25, 510–518. [Google Scholar] [CrossRef]
- Anand, K.; Roszik, J.; Gombos, D.; Upshaw, J.; Sarli, V.; Meas, S.; Lucci, A.; Hall, C.; Patel, S. Pilot Study of Circulating Tumor Cells in Early-Stage and Metastatic Uveal Melanoma. Cancers 2019, 11, 856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.J.; Diefenbacj, R.J.; Byrne, N.; Long, G.V.; Scolyer, R.A.; Gray, E.S.; Carlino, M.S.; Rizos, H. Circulating tumor DNA Reflects Uveal Melanoma Responses to Protei Kinase C Inhibition. Cancers 2021, 13, 1740. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.S.; Moore, L.R.; Joshi, P.; Goodin, M.; Zborowski, M.; Fleischman, A. Microfluidic chip for graduated magnetic separation of circulating tumor cells by their epithelial cell adhesion molecule expression and magnetic nanoparticle binding. J. Chromatogr. A 2021, 1637. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimonto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurements of protein using bicinchoninoc acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Ng, K.P.; Gugiu, B.; Renganathan, K.; Davies, M.W.; Gu, X.; Crabb, J.S.; Kim, S.R.; Rozanowska, M.B.; Bonilha, V.L.; Rayborn, M.E.; et al. Retinal pigment epithelium lipofuscin proteomics. Mol. Cell. Proteom. 2008, 7, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Crabb, J.W.; West, K.A.; Dodson, W.S.; Hulmes, J.D. Amino Acid Analysis. In Current Protocols in Protein Science; John Wiley & Sons, Inc.: Hobokin, NJ, USA, 2001; Volume 7. [Google Scholar] [CrossRef]
- Saikia, P.; Crabb, J.S.; Dibbin, L.L.; Juszczak, M.J.; Willard, B.; Jang, G.F.; Shiju, T.M.; Crabb, J.W.; Wilson, S.E. Quantitative proteomic comparison of myofibroblasts derived from bone marrow and cornea. Sci. Rep. 2020, 10, 16717. [Google Scholar] [CrossRef]
- Yuan, X.; Gu, X.; Crabb, J.S.; Yue, X.; Shadrach, K.; Hollyfield, J.G.; Crabb, J.W. Quantitative proteomics: Comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol. Cell. Proteom. 2010, 9, 1031–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollinger, K.E.; Crabb, J.S.; Yuan, X.; Putliwala, T.; Clark, A.F.; Crabb, J.W. Quantitative Proteomics: TGF-β2-Signaling in Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8287–8291. [Google Scholar] [CrossRef] [Green Version]
- Bollinger, K.E.; Crabb, J.S.; Yuan, X.; Putliwala, T.; Clark, A.F.; Crabb, J.W. Dexamethasone-Induced Proteomic Changes in Trabecular Meshwork Cells: Insights to the Molecular Basis of Steroid-Induced Glaucoma. Mol. Vis. 2012, 18, 2001–2011.72. [Google Scholar]
- Hultin-Rosenberg, L.; Forshed, J.; Branca, R.M.; Lehtio, J.; Johansson, H.J. Defining, comparing, and improving iTRAQ quantification in mass spectrometry proteomics data. Mol. Cell. Proteom. 2013, 12, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- R-Development-Team. R: A Language and Environment for Statistical Computing, R version 3.6.3; 29 February 2020; R-Development-Team: Vienna, Austria, 2019. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Waks, A.G.; Stover, D.G.; Guerriero, J.L.; Dillon, D.; Barry, W.T.; Gjini, E.; Hartl, C.; Lo, W.; Savoie, J.; Brock, J.; et al. The Immune Microenvironment in Hormone Receptor-Positive Breast Cancer Before and After Preoperative Chemotherapy. Clin. Cancer Res. 2019, 25, 4644–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Paeur, G.J.; Yue, X.; Narendra, U.; Sturgill, G.M.; Bena, J.; Gu, X.; Peachey, N.S.; Salomon, R.G.; Hagstrom, S.A.; et al. Assessing susceptibility to age-related macular degeneration with proteomic and genomic biomarkers. Mol. Cell. Proteom. 2009, 8, 1338–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welinder, C.; Ekblad, L. Coomassie staining as loading control in Western blot analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metastasizing | Non-Metastasizing | |
|---|---|---|
| Total pUM specimens | 53 | 47 |
| Total Proteins Quantified with ≥2 peptides | 3935 | 3934 |
| Average number proteins quantified per tumor | 2567 | 2541 |
| Proteins Elevated ≥ 1SD from Mean, adjusted p ≤ 0.05, imputation ≤ 20% | 232 | 224 |
| Proteins Decreased ≥ 1SD from Mean, adjusted p ≤ 0.05, imputation ≤ 20% | 206 | 201 |
| UniProt Accession | Gene Name | Protein (Sorted by Decreasing Protein Ratio) | Protein Ratio Met/NoMet | Adjusted p-Value | Frequency | |
|---|---|---|---|---|---|---|
| Metastasizing pUM | Non-Metastasizing pUM | |||||
| DE Proteins Most Abundant in Metastasizing pUM | ||||||
| P23381 | WARS1 | Tryptophan--tRNA ligase, cytoplasmic | 1.906 | 4.6 × 10−6 | 53 | 47 |
| P04439 | HLA-A | HLA class I histocompatibility antigen, A alpha chain | 1.818 | 1.6 × 10−6 | 53 | 47 |
| P61769 | B2M | Beta-2-microglobulin | 1.768 | 2.7 × 10−5 | 47 | 39 |
| P01903 | HLA-DRA | HLA class II histocompatibility antigen, DR alpha chain | 1.727 | 9.2 × 10−5 | 53 | 47 |
| Q03518 | TAP1 | Antigen peptide transporter 1 | 1.630 | 3.2 × 10−5 | 53 | 47 |
| P10321 | HLA-C | HLA class I histocompatibility antigen, C alpha chain | 1.624 | 1.8 × 10−5 | 52 | 46 |
| Q8IVF2 | AHNAK2 | Protein AHNAK2 | 1.616 | 8.8 × 10−4 | 44 | 36 |
| O95816 | BAG2 | BAG family molecular chaperone regulator 2 | 1.595 | 2.3 × 10−7 | 42 | 39 |
| P07686 | HEXB | Beta-hexosaminidase subunit beta | 1.589 | 1.9 × 10−4 | 53 | 47 |
| P33121 | ACSL1 | Long-chain-fatty-acid--CoA ligase 1 | 1.562 | 1.4 × 10−5 | 53 | 47 |
| P17931 | LGALS3 | Galectin-3 | 1.549 | 4.6 × 10−5 | 52 | 47 |
| P19971 | TYMP | Thymidine phosphorylase | 1.534 | 9.4 × 10−4 | 53 | 47 |
| P51810 | GPR143 | G-protein coupled receptor 143 | 1.530 | 4.6 × 10−4 | 53 | 47 |
| Q06210 | GFPT1 | Glutamine--fructose-6-phosphate aminotransferase [isomerizing] 1 | 1.505 | 3.8 × 10−5 | 53 | 47 |
| Q9H3G5 | CPVL | Probable serine carboxypeptidase CPVL | 1.486 | 7.4 × 10−4 | 53 | 47 |
| DE Proteins Least Abundant in Metastasizing pUM | ||||||
| P04792 | HSPB1 | Heat shock protein beta-1 | 0.726 | 1.4 × 10−5 | 53 | 47 |
| Q9UBI6 | GNG12 | Guanine nucleotide-binding protein G(I)/G(S)/G(O) subunit gamma-12 | 0.722 | 1.6 × 10−3 | 45 | 40 |
| Q9BZQ8 | NIBAN1 | Protein Niban 1 | 0.720 | 1.2 × 10−3 | 53 | 47 |
| Q8NC51 | SERBP1 | Plasminogen activator inhibitor 1 RNA-binding protein | 0.718 | 1.6 × 10−2 | 53 | 47 |
| P28161 | GSTM2 | Glutathione S-transferase Mu 2 | 0.710 | 5.4 × 10−4 | 53 | 47 |
| Q9P0M6 | MACROH2A2 | Core histone macro-H2A.2 | 0.708 | 1.1 × 10−5 | 47 | 39 |
| Q9NUJ1 | ABHD10 | Mycophenolic acid acyl-glucuronide esterase, mitochondrial | 0.707 | 6.8 × 10−7 | 53 | 47 |
| Q14240 | EIF4A2 | Eukaryotic initiation factor 4A-II | 0.697 | 2.3 × 10−7 | 53 | 47 |
| P05387 | RPLP2 | 60S acidic ribosomal protein P2 | 0.697 | 4.1 × 10−5 | 53 | 47 |
| P34913 | EPHX2 | Bifunctional epoxide hydrolase 2 | 0.688 | 8.4 × 10−5 | 53 | 47 |
| Q02252 | ALDH6A1 | Methylmalonate-semialdehyde dehydrogenase [acylating], mitochondrial | 0.681 | 1.3 × 10−6 | 53 | 47 |
| P21266 | GSTM3 | Glutathione S-transferase Mu 3 | 0.676 | 2.6 × 10−3 | 53 | 47 |
| P09211 | GSTP1 | Glutathione S-transferase P | 0.673 | 5.5 × 10−3 | 53 | 47 |
| Q02338 | BDH1 | D-beta-hydroxybutyrate dehydrogenase, mitochondrial | 0.672 | 1.4 × 10−5 | 50 | 44 |
| O75891 | ALDH1L1 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 0.604 | 4.6 × 10−6 | 46 | 42 |
| Uniprot Accession | Gene Nmae | Protein | Regression Coefficients | Odds Ratio | Protein Ratio Met/NoMet | Cell Surface Localization |
|---|---|---|---|---|---|---|
| P04439 | HLA-A | HLA class I histocompatibility antigen, A alpha chain | 0.436 | 1.547 | 1.818 | X |
| Q86UX7 | FERMT3 | Fermitin family homolog 3 | 0.028 | 1.029 | 1.419 | |
| P04062 | GBA | Lysosomal acid glucosylceramidase | 0.499 | 1.647 | 1.412 | |
| P67936 | TPM4 | Tropomyosin alpha-4 chain | 0.061 | 1.063 | 1.330 | |
| P21796 | VDAC1 | Voltage-dependent anion-selective channel protein 1 | 0.546 | 1.727 | 1.225 | X |
| A0FGR8 | ESYT2 | Extended synaptotagmin-2 | 0.497 | 1.643 | 1.216 | X |
| P13674 | P4HA1 | Prolyl 4-hydroxylase subunit alpha-1 | −0.310 | 0.733 | 1.211 | |
| P23368 | ME2 | NAD-dependent malic enzyme, mitochondrial | 0.050 | 1.051 | 1.201 | |
| Q15056 | EIF4H | Eukaryotic translation initiation factor 4H | 0.702 | 2.017 | 1.190 | |
| P50570 | DNM2 | Dynamin-2 | 0.131 | 1.140 | 1.175 | X |
| Q99829 | CPNE1 | Copine-1 | 0.072 | 1.075 | 1.174 | X |
| Q9HD67 | MYO10 | Unconventional myosin-X | 0.252 | 1.287 | 1.160 | X |
| P49748 | ACADVL | Very long-chain specific acyl-CoA dehydrogenase, mitochondrial | 0.072 | 1.075 | 1.154 | |
| Q00341 | HDLBP | Vigilin | 0.485 | 1.623 | 1.122 | X |
| P48729 | CSNK1A1 | Casein kinase I isoform alpha | 0.030 | 1.031 | 1.121 | |
| P53621 | COPA | Coatomer subunit alpha | 0.490 | 1.632 | 1.116 | |
| P11142 | HSPA8 | Heat shock cognate 71 kDa protein | 0.018 | 1.018 | 1.096 | X |
| P54920 | NAPA | Alpha-soluble NSF attachment protein | 0.050 | 1.051 | 1.095 | X |
| Q13616 | CUL1 | Cullin-1 | −0.178 | 0.837 | 0.903 | X |
| Q9BPX5 | ARPC5L | Actin-related protein 2/3 complex subunit 5-like protein | −0.102 | 0.903 | 0.895 | |
| P38606 | ATP6V1A | V-type proton ATPase catalytic subunit A | −0.124 | 0.883 | 0.885 | X |
| Q9BR76 | CORO1B | Coronin-1B | −0.110 | 0.896 | 0.874 | X |
| Q96TA1 | NIBAN2 | Protein Niban 2 | −0.448 | 0.639 | 0.864 | X |
| Q14344 | GNA13 | Guanine nucleotide-binding protein subunit alpha-13 | −0.213 | 0.808 | 0.844 | X |
| P01024 | C3 | Complement C3 | −0.124 | 0.883 | 0.840 | X |
| Q8N1G4 | LRRC47 | Leucine-rich repeat-containing protein 47 | −0.391 | 0.677 | 0.838 | |
| Q14624 | ITIH4 | Inter-alpha-trypsin inhibitor heavy chain H4 | −0.380 | 0.684 | 0.836 | X |
| Q9NXF1 | TEX10 | Testis-expressed protein 10 | −0.512 | 0.599 | 0.802 | |
| P62899 | RPL31 | 60S ribosomal protein L31 | −0.094 | 0.911 | 0.795 | |
| Q96I99 | SUCLG2 | Succinate--CoA ligase [GDP-forming] subunit beta, mitochondrial | −0.254 | 0.775 | 0.789 | X |
| Q9BZQ8 | NIBAN1 | Protein Niban 1 | −0.095 | 0.910 | 0.720 | X |
| P28161 | GSTM2 | Glutathione S-transferase Mu 2 | −0.171 | 0.843 | 0.710 |
| TCGA Coding mRNA | Laurent Coding mRNA | TCGA Transcripts DA SCNA Cluster 3 vs. 4 | TCGA + Laurent Transcripts DA SCNA Cluster 3 vs. 4 | TCGA Transcripts Up in mRNA Cluster 3 vs. 4 | TCGA Transcripts Down in mRNA Cluster 3 vs. 4 | |
|---|---|---|---|---|---|---|
| Number transcripts * | 12,319 | 13,142 | 591 | 510 | 338 | 2172 |
| Transcripts Identified at protein level * | 3433 | 3524 | 133 | 128 | 109 | 373 |
| Fraction of transcript detected as proteins | 27.9% | 26.8% | 22.5% | 25.1% | 32.2% | 17.2% |
| Total DE proteins | 378 | 364 | 20 | 19 | 18 | 28 |
| DE proteins Elevated in Mets | 181 | 181 | 17 | 17 | 5 | 18 |
| DE proteins Decreased in Mets | 197 | 183 | 3 | 2 | 13 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, G.-F.; Crabb, J.S.; Hu, B.; Willard, B.; Kalirai, H.; Singh, A.D.; Coupland, S.E.; Crabb, J.W. Proteomics of Primary Uveal Melanoma: Insights into Metastasis and Protein Biomarkers. Cancers 2021, 13, 3520. https://doi.org/10.3390/cancers13143520
Jang G-F, Crabb JS, Hu B, Willard B, Kalirai H, Singh AD, Coupland SE, Crabb JW. Proteomics of Primary Uveal Melanoma: Insights into Metastasis and Protein Biomarkers. Cancers. 2021; 13(14):3520. https://doi.org/10.3390/cancers13143520
Chicago/Turabian StyleJang, Geeng-Fu, Jack S. Crabb, Bo Hu, Belinda Willard, Helen Kalirai, Arun D. Singh, Sarah E. Coupland, and John W. Crabb. 2021. "Proteomics of Primary Uveal Melanoma: Insights into Metastasis and Protein Biomarkers" Cancers 13, no. 14: 3520. https://doi.org/10.3390/cancers13143520