|
To view this email as a web page, click here. |
 |
 |
|
Welcome
We have a de novo sequencing competition to exercise your brain cells over the holiday period. The MS/MS spectrum of a single peptide can be downloaded here as an MGF peak list. The peptide may be modified and you shouldn't assume it is tryptic.
You can figure out the sequence in any way you like, but the way to approach it using Mascot Distiller is described in our October blog article. Email your solution to newsletter@matrixscience.com to arrive before 12 noon GMT on December 24th. From the fully correct solutions, we will draw three winning entries. For each winner, we will donate $100 to the WHO COVID-19 Response Fund and the names of the winners will be listed in a roll of honour in our January newsletter (unless your entry says you prefer to remain anonymous).
We have some tips and tricks concerning sequence database management.
This month's highlighted publication has a new approach to identify and locate ADP-ribosylation of peptides and proteins.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month highlights a review paper full of practical tips for database searching.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
The minutiae of database management
Though the major inputs into a database search are only sequence databases and mass spectrometry data, there are some common problems that sometime arise and cause frustration. Here are a few suggestions on how to minimize their impact on your work.
- New database test search can time out - Increase the MonitorTestTimeout in the Configuration Editor.
- If you are working with non-lab species, like barnyard animals, we recommend setting up a custom proteome database using the Uniprot proteome template.
- After activating a new database, if you see an error reporting the accession string as over 50 characters, go back to the configuration details and choose a better parse rule.
- There is a soft limit on the length of individual sequences and if you exceed the default, you will see an error. Edit the MaxSequenceLen option
- When searching nucleic acid databases, Mascot Server will perform a 6-frame translation on the fly. Some third-party software clients do not support searching of nucleic acid databases, so you will need to submit the search using the web browser search form or Mascot Daemon.
Go here to read more details about these and other tips.
|

|
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Gas-Phase Fragmentation of ADP-Ribosylated Peptides: Arginine-Specific Side-Chain Losses and Their Implication in Database Searches
Peter M. Gehrig, Kathrin Nowak, Christian Panse, Mario Leutert, Jonas Grossmann, Ralph Schlapbach, and Michael O. Hottiger
Journal of the American Society for Mass Spectrometry published online November 3, 2020
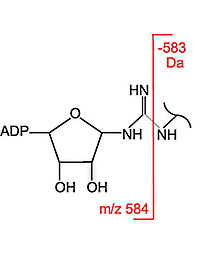
Identifying the modification and site localization of ADP-ribosylated peptides is difficult due to the instability of the modified peptides and the large number of possible amino acid acceptor sites. The authors used samples from HeLa cells and wild-type mice skeletal muscle tissue that were trypsin digested and enriched with ADP-ribose-binding macro domain proteins. LC-MS/MS with HCD survey scans as well as HCD-triggered EThcD MS/MS provided rich spectra with fragmentation of the ADP-ribose moiety or the peptide backbone.
The authors customised the ADP-Ribosyl modification to include the loss of the 583.08 Da fragment from b and y ions in database searches. This increased the assignments of ADP-ribosylated peptides by at least 30%. Both peptide identification scores and PTM localization scores were improved because of the higher counts of identified sequence-specific fragment ions.
The HCD fragmentation method and the inclusion of the 583.08 Da neutral loss are mainly recommended for samples rich in ADP-ribosylated arginine. The more labile O-glycosidic linkages between ADP-ribose and serine, glutamic acid, or aspartic acid make localization of these modifications difficult since this was the major cleavage site and provided few diagnostic ions.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|