|
To view this email as a web page, click here. |
 |
|
Welcome
Please join us at ASMS for our annual User Meeting on Monday June 6.
Mascot Distiller processing options for peak picking allow you to choose the optimum approach.
This month's highlighted publication shows how crosslinking can help unravel complex interactions.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month discusses the glycan entries in Unimod
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
 |
|
Matrix Science ASMS User Meeting
Join us for breakfast at ASMS in Minneapolis and learn about the exciting applications of database searching as well as how to get the most out of Mascot.
There is no charge for attending this meeting, but advance registration is required. Breakfast will be provided.
Monday 6 June, 7:00 am - 8:00 am
Identification and Quantification of Histone Methylation in Cancer Cells
presented by Dr Roland Annan, GlaxoSmithKline
NCBIprot, mzIdentML 1.2 and other improvements in Mascot Server 2.8.1
presented by Ville Koskinen, Matrix Science
Fractionated LFQ in Mascot Distiller 2.8.2
presented by Patrick Emery, Matrix Science
|
 |
|
|
 |
|
|
|
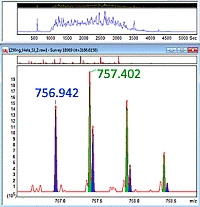
Peak picking with Mascot Distiller
Mascot Distiller contains a number of sets of processing options to provide reasonably good peak picking for each of the vendors' raw files. These are good starting points, and will allow you to further optimize the settings if you want even better results. Here we will take a look at the two main options for processing Thermo data.
In both cases, Distiller will use profile data for peak detection in the MS scans, which is required if you want to carry out quantitation using survey scan based methods such as label-free, SILAC etc.
The main point of difference between the two sets of processing options is that the "default" settings will accept centroid values for peaks in MS/MS scans if this is what the raw data contains, while the "prof_prof" processing options will always uncentroid these types of scans and then carry out its own peak picking. This will also provide the fragment ion charge states which are needed for de novo sequencing, and can be used to de-charge the peaklists, which is important if you are doing top- or middle- down experiments.
As a general guideline, if your MS/MS scans are saved as profile data you should use the "prof_prof" settings as your starting point for peak picking. This will provide many more significant matches in a similar time to the "default" method. However, for MS/MS scans saved as centroids, the "default" processing option will yield nearly the same quality matches as the "prof_prof" option but without the time penalty incurred by the uncentroiding of the data.
Go here to read more recommendations about processing options.
|
|
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
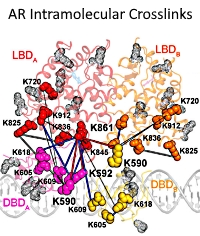
Allosteric interactions prime androgen receptor dimerization and activation
Elizabeth V. Wasmuth, Arnaud Vanden Broeck, Justin R. LaClair, Elizabeth A. Hoover, Kayla E. Lawrence, Navid Paknejad, Kyrie Pappas, Doreen Matthies, Biran Wang, Weiran Feng, Philip A. Watson, John C. Zinder, Wouter R. Karthaus, M. Jason de la Cruz, Richard K. Hite, Katia Manova-Todorova, Zhiheng Yu, Susan T. Weintraub, Sebastian Klinge, and Charles L. Sawyers
Molecular Cell, 82 1-11 (2022)
The androgen receptor (AR) is a nuclear receptor that regulates gene expression, and its aberrant activity can lead to pathologies such as prostate cancer and androgen insensitivity syndrome. The AR contains an intrinsically disordered N-terminal domain, a DNA-binding domain (DBD), a flexible hinge, and a ligand binding domain (LBD). The authors have sought to unravel AR signaling, which is controlled through a variety of intramolecular and external interactions.
They utilized protein crosslinking to gain insight into the molecular features that govern the AR's activation. The AR complex was generated by combining AR, the oncoprotein ERG, and the DNA androgen response elements. The complex was crosslinked with disuccinimidyl sulfoxide (DSSO) for 1 hour, quenched, and then separated by ultracentrifugation. The complex was further separated on SDS-PAGE, excised, digested, and analyzed by LC-MS/MS. The Mascot cross-linking feature for DSSO was used for the sample searches, and data were imported into xiView for visualization of the cross-linked sites.
The highest scored crosslinks were at the interface between the AR DBD and the LBD, supporting the authors' domain docking and structural observations. Additionally, the two most enriched cross-linked lysines in the DBD comprise part of the "lever arm", which has been thought to mediate interdomain allostery via its flexibility.
|
 |
|
|
|
|
|
|
|
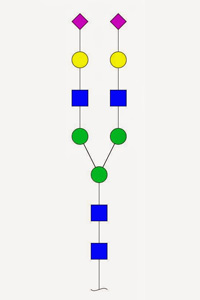
Mascot Tip
Glycopeptide analysis isn't easy. Many sugars are large and heterogeneous. In CID / HCD, the glycan tends to fragment at the expense of the peptide, so that sequence ions from the peptide are weak, and the entire glycan is lost, making site analysis impossible. ETD is preferred since it keeps the glycan intact and in situ.
Unimod currently includes 450 entries for sugars, intended to cover all naturally occurring N-linked and O-linked glycans with masses of 2 kDa or less found on human proteins. The S and T specificities for O-linked glycans are grouped, so that they can be selected as a single modification, and all of them have neutral losses defined of zero, for ETD, and the complete glycan, for CID / HCD.
This is very convenient if glycopeptides are of interest. If not, be aware that the default behaviour for an error tolerant search is to test every modification in the local Unimod file. If glycopeptides are either not expected or not of interest, especially the larger ones, you can speed up the search and make it more specific by excluding these potential modifications. The way to do this is explained in an earlier newsletter.
|
|
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
