You’ve got to pick a precursor or two: Precursor detection in Mascot DIA
We’re working on a universal spectrum-centric solution for the analysis of DIA data, called Mascot DIA. A preview was presented at our ASMS 2025 Breakfast Meeting, the slides from which are free to download. Last month’s blog article outlines the spectrum-centric search approach for DIA data and compares it to the more common peptide-centric approach. One thing that is essential for the spectrum-centric approach is accurate precursor detection, so how are we tackling this in Mascot DIA?
Precursor detection in Mascot DIA is handled by Mascot Distiller, as outline in the figure below:
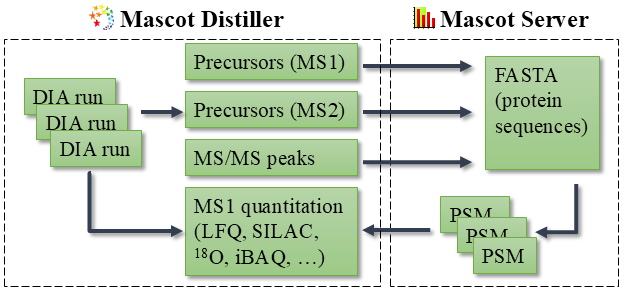
Figure 1: The Mascot DIA workflow. Precursor detection and peak picking is carried out in Mascot Distiller and the peaklists searched using a Mascot Server database search. Results are imported back into Mascot Distiller for MS1 based quantitation
Precursor detection for DIA data in Mascot Distiller 3
Precursor detection for DIA data in Mascot Distiller is a two stage process. Initially the software looks in the survey scans. If fails to find sufficient precursors in the survey scan it uses a novel algorithm to look for precursors within the MS/MS spectrum.
The main advantage of this approach is that precursor detection, and therefore peptide identification, is not dependent on any library of precursor masses and associated retention times, whether experimentally or computationally derived. This allows the software to potentially detect peptides with any post-translational modifications and from any enzyme digest supported by Mascot Server.
Stage 1: Look in the survey scans
As with a standard DDA run, precursor detection for DIA runs in Mascot Distiller starts from the MS1 survey scans. The survey scan is split into the isolation windows used in the DIA experiment and precursor masses selected within each window and assigned to the associated MS/MS scan. This uses the same algorithm as is used for precursor detection of DDA data.
For DIA data the assumption is that every scan is chimeric and comes from multiple precursors, so if Distiller fails to find more than 1 precursor in the survey scan for any given isolation window, it switches to looking for additional precursor signals in the child MS/MS scan. This is new functionality being added to Mascot Distiller 3 to support DIA runs.
Stage 2: Look in the MS/MS scans
You often see unfragmented precursor signal in the MS/MS scans from DIA datasets, so one option would be to look in the isolation window region of the MS/MS scan and pick out potential unfragmented precursors from there. In practice we found this approach isn’t very reliable for a number of reasons. One issue is that if the precursor is missing from the survey scan, it is almost by definition a low intensity precursor, which means you’re less likely to find any unfragmented precursor signal in the MS/MS scan so you’re likely to miss it, or it will be swamped by other noise.
Another other issue is that if the ‘raw’ data are actually saved as centroids, as is often the case for data from Thermo instruments like the Orbitrap, Mascot Distiller won’t detect the charge states of fragment ions without uncentroiding the spectrum to convert it back into profile data, which can significantly extend the processing time.
Instead, Mascot Distiller uses a novel algorithm to infer precursor masses within the isolation window based on pairs of complementary ions (e.g. b- and y- ions).
- Add together the masses of pairs of fragment ions from the MS/MS spectrum. This gives you a potential precursor MH+ mass plus an additional proton. Take away the mass of the additional proton – does the MH+ precursor mass fall into the isolation window? If it does, add the MH+ mass to a long list of possible precursors.
- If the mass is greater than the precursor window, convert it to a range of default charge states specified in the processing settings (e.g. 2+, 3+ etc). If any of those fall into the isolation window, add the precursor to the long list.
- Repeat steps 1 and 2 until the MS/MS spectrum is covered.
- Now rank the precursors in the long list by the number of times the precursor was observed by different fragment ion pairs and by coverage of the spectrum and take up to the top N precursors and assign to the spectrum (N is 10 by default).
- It is data driven from the MS/MS spectrum – you don’t need a library of precursor masses and associated retention times.
- It doesn’t matter what variable (or static) modifications are present on the peptide and it doesn’t matter what enzyme was used in the digestion step (if one was used at all).
- It determines precursor mass and charge even from centroided data.
- It is largely independent of fragmentation efficiency – so long as the peptide has fragmented well enough to give a few complementary ions it can determine the precursor mass.
- It works with any complementary ion series, b- and y-, c- and z- etc.
Example of MS/MS based precursor detection
Here’s an example from a match from a DIA dataset where the precursor m/z and charge state was determined from the MS/MS scan like this. The match is shown in figure 2 below:
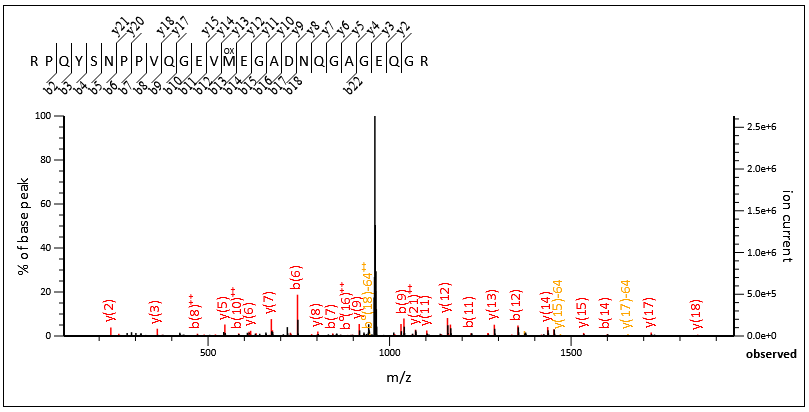
Figure 2: MS/MS Fragmentation of RPQYSNPPVQGEVMEGADNQGAGEQGR
As you can see we have good coverage across the entire peptide. If we zoom into a region around 1150-1750, we have a number of complementary pairs of ions here; b12+y15, b13+y14, b14+y13:
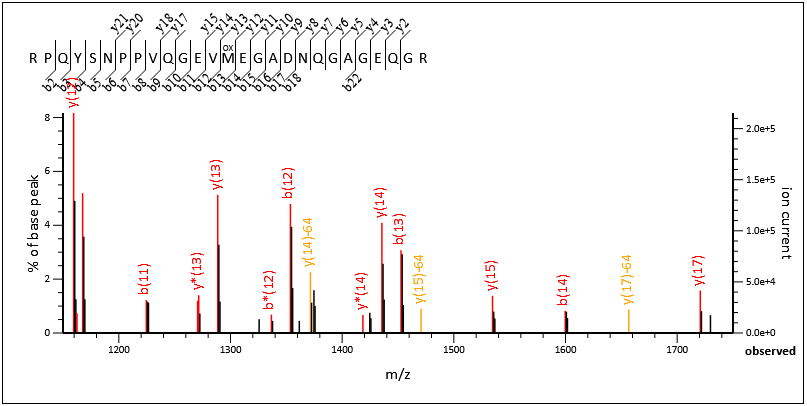
Figure 3: Zoomed in region 1150-1750 containing a number of complementary ion pairs.
The calculated mass and charges of these pairs are shown in table 1 below:
| Pair | m/z b- ion | m/z y- ion | MH+ plus proton | 1+ | 2+ | 3+ |
|---|---|---|---|---|---|---|
| b12+y15 | 1353.652 | 1534.6479 | 2888.3 | 2887.293 | 1444.15 | 963.1024 |
| b13+y14 | 1452.7212 | 1435.5948 | 2888.316 | 2887.309 | 1444.158 | 963.1078 |
| b14+y13 | 1599.7555 | 1288.5535 | 2888.309 | 2887.302 | 1444.155 | 963.1054 |
Table 1: Example precursor calculation from three pairs of complementary ions
The isolation window for this MS/MS scan was 956.687-964.687, so the precursor must have an m/z of 963.1 and a charge state of 3+ to fit into that region. The actual calculated precursor from Distiller is 963.10696,3+ and the precursor error is 1.37ppm. Note that although there are strong unfragmented precursor peaks in the isolation window region of the MS/MS scan (as can bee seen in figure 2), none of those match this precursor. Figure 4 below shows the counts of b- y- complementary pairs for the different precursor masses in the isolation window identified from the MS/MS scan. In this case, our precursor m/z 963.10696 3+ is a clear outlier:
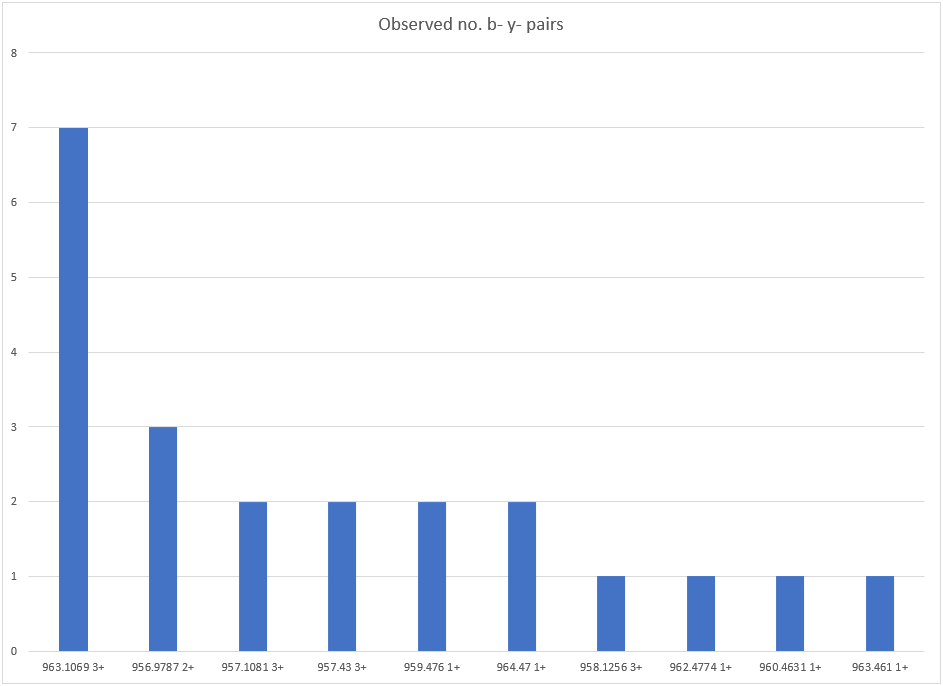
Figure 4: Barchart showing the number of complementary b- y- ion pairs for the various precursors identified from the MS/MS peaklist.
Reference
- Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De Novo Peptide Sequencing via Tandem Mass Spectrometry. Journal of Computational Biology 6(3/4):327-342. DOI: 10.1089/106652799318300
Keywords: chimeric spectra, DIA, Mascot Distiller, peak picking