30 years of Sequence Tags
This year we are celebrating 30 years of sequence tags. The technique was developed by Matthias Mann and Matthias Wilm while in the Protein and Peptide group at the European Molecular Biology Laboratory (EMBL). EMBL is itself celebrating its 50th Anniversary and has been an important center for science over that period. The sequence tag paper was published in 1994, the same year as the micro electrospray paper, also from the Mann lab, and both techniques go hand in hand as far as enabling the identification of peptides and proteins in that time period.
The original paper has a great figure describing a sequence tag:
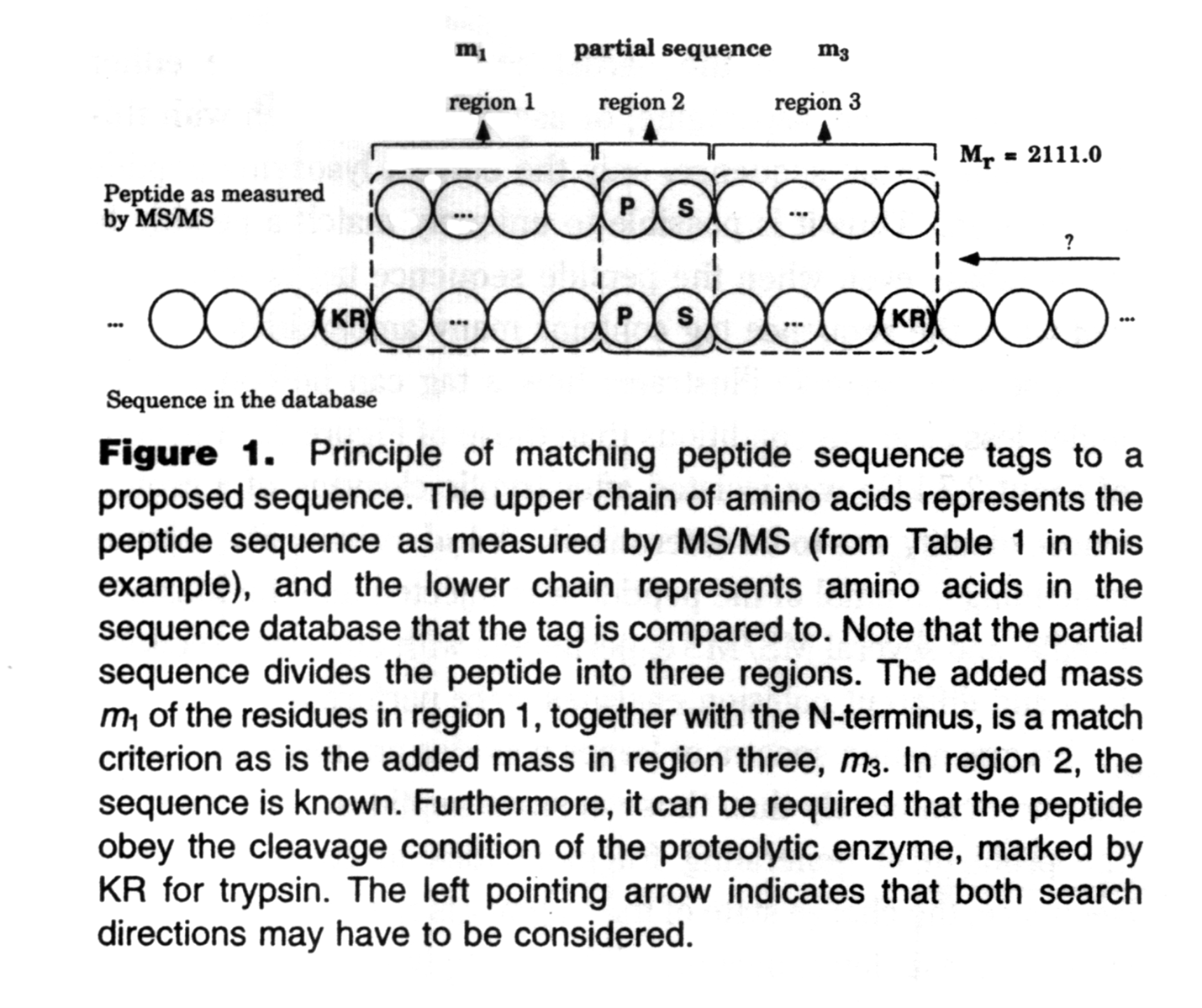
The first version of Mascot Server supported sequence tags, one of several techniques that use semi-annotated spectra or sequence information to identify peptides. There were a number of publicly accessible search engines for sequence tags and related searches, but the only two that still offer the feature that I know of are ProteinProspector and Mascot Server. If you want to know about how to call a sequence tag and what to do with it, I recommend reading the Sequence Queries tutorial.
I was fortunate enough to be working in the Protein and Peptide group at EMBL as a peptide chemist in 1994 and can add a few more details about the developments at the time. Digested proteins were desalted in a pulled glass needle packed with C18 column material and spun into a second pulled needle that was used as the electrospray source for a Sciex API III mass spectrometer. The breakthrough brought by the micro electrospray, later renamed the nanoelectrospray, was that a single µL of sample now lasted 40 minutes or more rather than the minute or two with the electrospray source the instrument was originally equipped with. During the acquisition, peptides were selected for MS/MS manually and as soon as enough information was collected, the next peptide was selected, etc. This was quite an exciting process as the sample was being consumed so it was a race against the clock.
At the time the lab did not have any software to call the sequence tags, so the data was processed manually. To speed up the process, an AppleScript formatted the spectrum into windows of fixed size in Daltons and printed them, normally two to four pages per peptide. There was a ruler with the amino acid masses marked on it and printed on to a transparent acetate typically used with overhead projectors. Starting from a large peak, you would use the ruler to select the next peak that matched an amino acid difference and build a ladder of multiple amino acids that were hopefully in the same ion series. Once you had 3 or more amino acids, you would use the two terminal masses to complete the tag and enter it into the PeptideSearch sequence tag search engine, also written by Matthias Mann and the lab. As researchers became more experienced, they would often read off a tag while the spectra was being acquired and submit a search before selecting the next target peptide.
Even at the time of the sequence tag publication, it was noted that there were methods to search uninterpreted data that were more suited for automation. Once the LCMS/MS methods were established sequence tags became essentially obsolete.
The actual process of creating a manual sequence tag in Mascot Distiller is very similar to doing so back in the day. Here is some Bruker timsTOF data. I’ve selected a peak at 1279 m/z and extended from there to create a tag:
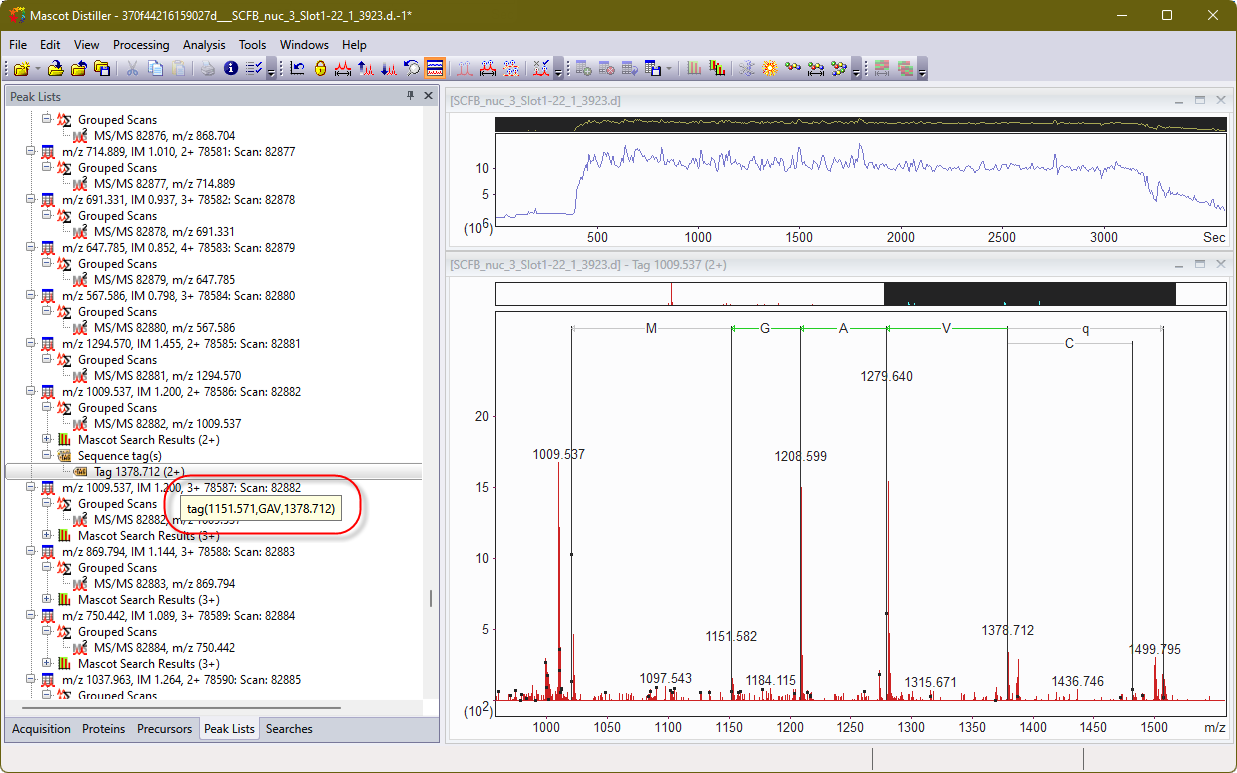
Given the manual nature and difficulty in correctly calling a sequence tag, why does Mascot Server still support the analysis method? The answer is error tolerant sequence tags. An error tolerant tag (etag) allows for an unsuspected modification or a small difference in the sequence, a single nucleotide polymorphism for example. When the etag is searched, the peptide molecular weight constraint is relaxed and the fragment ion mass values must fit one of two possibilities. Either both values are unchanged or both values are shifted by the same amount as the peptide mass.
Rather than calling the sequence tag manually, you can also use the Mascot Distiller search toolbox de novo algorithm to create solutions for spectra that did not have a significant match in a standard uninterpreted search.
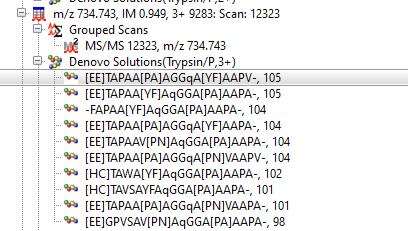
You can then search these de novo solutions as etags. Some of the ones generated from the above de novo solutions are shown here:
2201.207624 from(2202.214900,1+) title(9283: Scan 12323 (rt=11.8765, p=9379) index(0) etag(1944.16966,TAP,1674.99221) etag(1772.04349,PAA,1532.91628) etag(1293.79272,GG[Q|K],1051.56527)
There are a number of similar scoring search results. This one from the “PAA” tag looks interesting and fairly easy to explain:
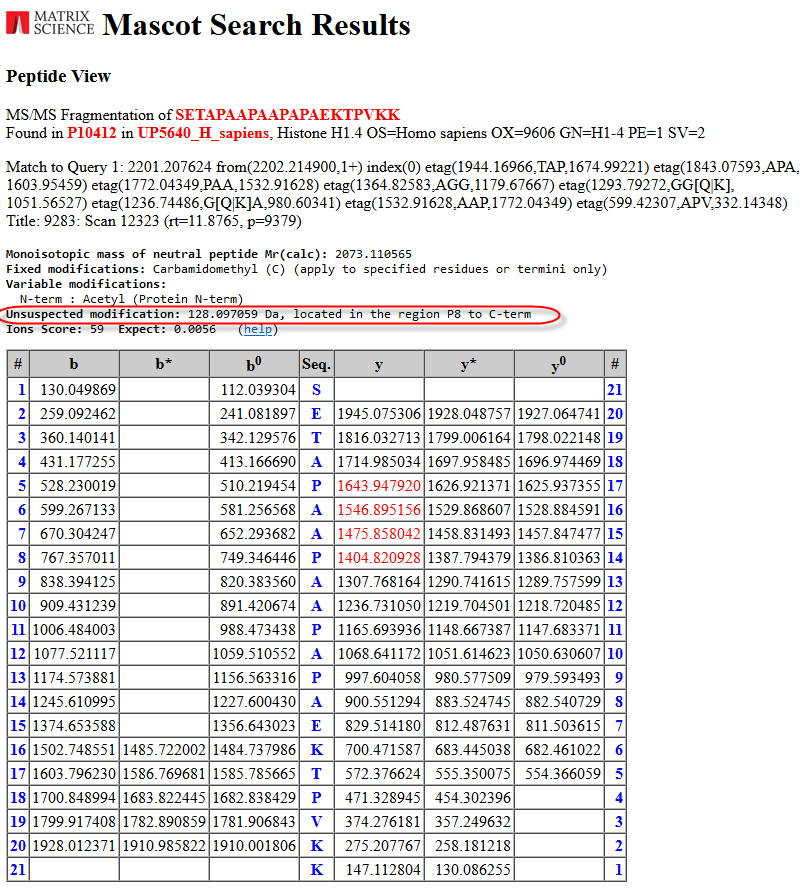
The unsuspected modification with a mass of 128.09Da, an addition of a Lysine, is located in the region P8 to the C-terminal. All the masses of the tag have been shifted by the modification mass. From the Protein View report we can indeed see that the next amino acid in the protein is a Lysine:
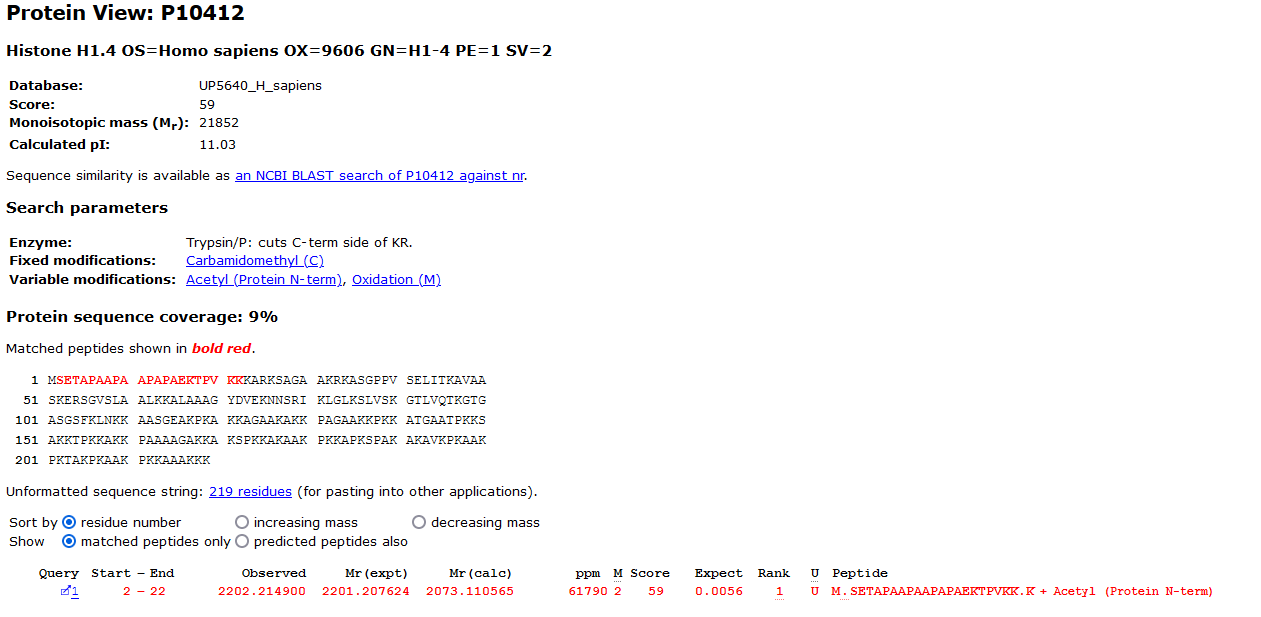
Looking back at the spectra, I believe it was not identified in the original MS/MS search because it is chimeric and there are a lot of large unassigned peaks. A change in peak picking settings may help identify it future searches.
Error tolerant sequence tag searches are the last step in a search strategy after standard searches, error tolerant searches and de novo sequencing. If you have a boutique problem, etags are still a useful technique to have in the toolbox.
Do you remember when sequence tags were introduced and were you selecting peptides for sequencing by hand? Post a memory in the comments.
Keywords: anniversary, de novo, error tolerant, history, Mascot Distiller