Higher accuracy for oxidation profiles
Earlier this year, Reest et al. described a quantitation workflow based on light and heavy iodoacetamide labels, which they named SICyLIA [1]. The workflow was used for analysing cysteine oxidation levels between wild-type and redox-stressed mouse cells.
At its core, SICyLIA is a precursor protocol with two components, light and heavy, where the labels are simply light and heavy carbamidomethyl on cysteine (CAM). However, there’s a twist: reversibly oxidised cysteine thiols are reduced and blocked with N-ethylmaleimide (NEM) after pooling. Light and heavy peptides with more than one cysteine may well have NEM-blocked cysteines, and there may be peptides carrying only a NEM.
Reest et al. note that 15-16% of peptides in the study have multiple cysteines. Those with both a label and a NEM-blocked cysteine (3-4%) were omitted from quantitation analysis due to software limitations. But it’s easy to define a quantitation method for Mascot Distiller that accounts for NEM-blocking.
CAM quantitation method
In most Precursor quantitation methods, light and heavy components are modelled as exclusive modifications, which are treated like fixed modifications during the search. This means all potential sites for labelling must be identically modified.
However, in SICyLIA, we want to allow zero or more NEM-blocked cysteines. Easily done: define the components using variable modification groups instead of exclusive mods. This allows Mascot to match peptides in all eight cases (unmodified; NEM; light; heavy; light + NEM; heavy + NEM; light + heavy; light + heavy + NEM). Distiller assigns a peptide to the light component if it carries at least one light CAM, and to the heavy component if it carries at least one heavy CAM. If the peptide has both light and heavy CAM, it correctly fits neither component.
Configuration must be done on the Mascot Server side.
First, add the heavy Carbamidomethyl as a locally defined
modification in the Mascot Configuration Editor. It’s best to
make a copy of Carbamidomethyl and change the composition to the one
given in the paper,
13C(2) 2H(2) H N O, because this way all the specificities
are carried over.
In the Mascot configuration editor, create a new quantitation method “SICyLIA CAM forward [MD]“:
- Set method to precursor.
- Create component “light”. Add a mod group containing Carbamidomethyl (C) from Unimod.
- Create component “heavy”. Add a mod group containing heavy Carbamidomethyl (C). Define impurity corrections that put 99% of intensity on 13C and deuterium and 1% on 12C and hydrogen (following impurity levels reported in the paper).
- Set mode to variable in all mod groups, and tick required.
- In the Method tab, create a modification group “NEM”. Add the variable modification N-ethylmaleimide (C) from Unimod.
- Add a ratio L/H with denominator “heavy” and numerator “light”. Set protein ratio type to median.
The method is named “forward”, because the authors used a label-swap replication strategy. Forward replicates mix untreated heavy samples with treated light samples, so the ratio of interest is L/H. For reverse replicates, copy the method and invert the ratio definition.
Correct settings for integration and quality are important but not relevant to the topic at hand. For more detail, see the Distiller help (Quantitation Toolbox -> Method Configuration – Precursor).
More quantifiable peptides
To test the quantitation method, we downloaded fractions 2-21 for forward replicate 1 of the Fh1 cell model for the CAM-labelled case from PRIDE project PXD006372, 240616_SEL_HF_LEZ_R12_160616_WT-FH_r01_fwd_f*.raw. (Fraction 1 was excluded, because the raw file’s title indicates it is actually a reverse replicate.)
The raw files were peak picked in Mascot Distiller as an independent multi-file project. All searches were run as decoy searches using the SICyLIA CAM forward [MD] quantitation method against SwissProt with Mus. musculus taxonomy at 20ppm precursor tolerance and 20ppm fragment tolerance. Reest et al. specify Oxidation (M) and Acetyl (N-term) as the only variable modifications, so we’ll use the same.
The search results show a number of additional matches to light or heavy NEM-blocked peptides, as well as unlabelled NEM-blocked peptides. Among the 214,755 peptide matches at 2% FDR, the modification counts are:
| Modification | Site | Above thr. |
|---|---|---|
| Carbamidometdyl:2H(2)13C(2) | C | 23966 |
| Carbamidomethyl | C | 21180 |
| Oxidation | M | 13834 |
| Acetyl | N-term | 9652 |
| Nethylmaleimide | C | 3433 |
The screenshot below illustrates the NEM effect on quantitating just one protein hit, ITB1_MOUSE.
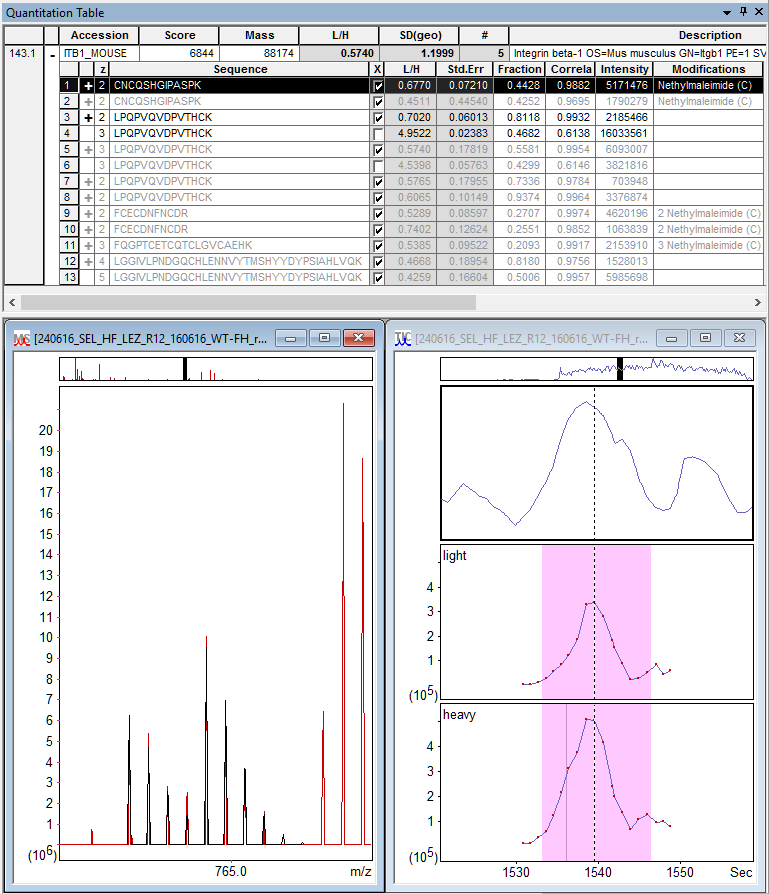 Figure 1: Distiller quantitation results for ITB1_MOUSE (Integrin beta-1).
Top: table of quantifiable peptide matches. Bottom: predicted isotope peaks
of CNCQSHGIPASPK + NEM (C) fitted to the survey scan and XICs for the two components.
Figure 1: Distiller quantitation results for ITB1_MOUSE (Integrin beta-1).
Top: table of quantifiable peptide matches. Bottom: predicted isotope peaks
of CNCQSHGIPASPK + NEM (C) fitted to the survey scan and XICs for the two components.
You can see the protein has NEM-blocked matches to three new quantifiable peptide sequences (CNCQSHGIPASPK, FCECDNFNCDR, FQGPTCETCQTCLGVCAEHK). The number of ratio measurements increases from 8 to 13. Repeat measurements of the same sequence and modification state are combined using the same ratio type as protein ratio, in this case the median, so the protein ratio sample size increases from 2 to 5. In other cases, even if the protein ratio sample size were to stay the same, repeat measurements improve the peptide ratio estimate, which in turn improves protein ratios.
References
- Jiska van der Reest, Sergio Lilla, Liang Zheng, Sara Zanivan and Eyal Gottlieb: “Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress”. Nature Communications 9:1581 (2018) doi:10.1038/s41467-018-04003-3
Keywords: carbamidomethyl, iodoacetamide, Mascot Distiller, quantitation