The plus one dilemma
There are several common modifications that can add approximately 1 Da to a peptide mass. Even if you have high accuracy data, it can sometimes be difficult to figure out which one is correct.
| Delta | |
|---|---|
| Lys->Glu substitution | 0.947630 |
| Leu->Asn or Ile->Asn substitution | 0.958863 |
| Deamidation at N or Asn->Asp substitution | 0.984016 |
| Deamidation at Q or Gln->Glu substitution | 0.984016 |
| Citrullination at R | 0.984016 |
| Label:15N(1) | 0.997035 |
| 13C peak of isotopic envelope | 1.003355 |
The level of 13C peptide masses in a peak list depends strongly on the peak picking software. Mascot Distiller does well because it fits the experimental isotope distribution to a calculated distribution for averagine. Usually, this will lock-in on the 12C peak. But, it is not infallible, and may fail on very weak peaks, where the relative intensities across the isotopic envelope are badly distorted.
For a typical tryptic peptide of mass 1500 Da, the difference between deamidation and picking the 13C isotope peak is 13 ppm. If you have low ppm accuracy for the precursor, you should be able to discriminate between these two by peptide molecular mass. If not, you depend on the quality of the MS/MS match and the accuracy of the MS/MS fragment masses. For deamidation, the fragments that include the modified residue are +1. If the wrong isotope peak has been picked, none of the fragments are +1. Unless the fragment mass accuracy is poor, so that the tolerance has to be set very wide, this should result in a score difference between the two possible matches.
Here is a good example where the tolerance is wide enough to allow matches to both possibilities. Deamidation on Q14 gets a score of 94 and there are matches to both b and y fragments containing the modified residue. If the deamidation is moved to other locations, the score drops off. The 13C match loses the y ion matches and the score drops to 53.
It helps if the MS/MS data have been de-isotoped. If not, there are +1 fragment matches available from the 13C isotope peaks for the fragments. This doesn’t destroy the score difference, because these will mostly be weaker than the 12C, forcing Mascot to select more peaks, which reduces the score. But, it will narrow the gap. A more serious limitation is when the modified residue is near the amino terminus and the predominant fragments are y ions, so that there are few or no +1 peaks to be observed. (Or, vice versa, with the modification near the carboxy terminus but a spectrum full of b ions.)
No matter how good your mass accuracy, you cannot use peptide molecular mass to distinguish deamidation from citrullination; they have an identical mass shift. This was the subject of a paper from the Kuster lab, which we featured in our May 2018 newsletter. For peptides where both possibilities existed, true citrullination was validated by a combination of site localisation, the neutral loss of isocyanic acid, the immonium ion of citrulline, and comparison with reference spectra of synthetic citrullinated and deamidated peptides. The authors also noted that trypsin will not cleave after citrulline, so that PSMs with modified C-terminal arginine can be rejected.
If you have ever run an error tolerant search, you may have seen matches to peptides modified by Label:15N(1). This is somewhat misleading. Label:15N(1), Label:15N(2) and Label:15N(3) are only present in Unimod because some software uses these modifications for matching peptides that have been metabolically labelled with 15N. They are not used by Mascot, which calculates metabolic mass shifts direct from the elemental compositions of the residues.
The reason that Label:15N(1) is seen so often in error tolerant results is that it has 14 specificities, while deamidation has only 2. For non-ideal spectra – i.e. most spectra with limited signal/noise – these additional degrees of freedom mean that Label:15N(1) can easily get a slightly better score than Deamidation. For example:
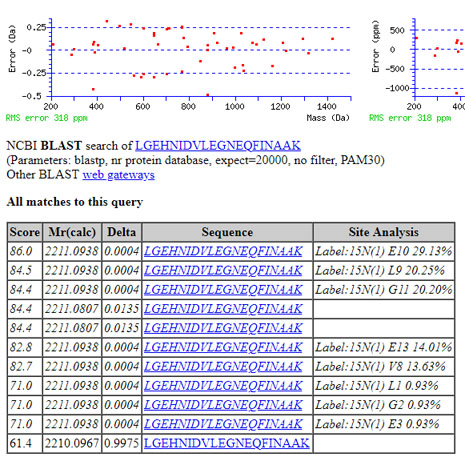
(The 4th and 5th rank matches are deamidation and the lowest rank match is for 13C.) This confusion can be avoided by excluding some or all of the isotope label modifications in Unimod from error tolerant searches. The way to do this is described in the Mascot tip in our November 2017 newsletter.
Keywords: 13C, 15N, citrullination, deamidation, error tolerant