The three Ds: Diethylation, Dimethylation and the Deuterium effect
Dimethylation
Mascot Server and Mascot Distiller support a number of precursor intensity based quantitation techniques using a number of different techniques and chemistries. The software also allows you to flexibly define your own methods, allowing you to implement quantitation protocols which we have not supplied ‘out of the box’.
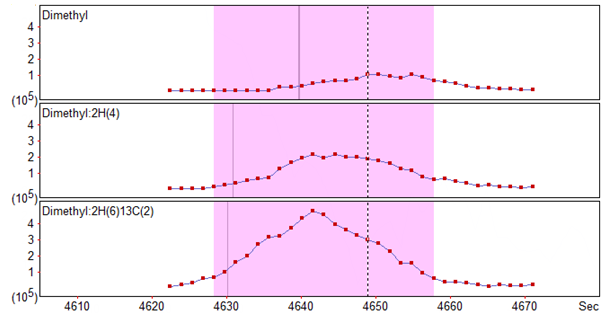
One isotope labelling method which Distiller supports ‘out of the box’ is the 3-plexed dimethylation method, which involves the labeling of N-terminus and Lysines of samples with light, medium and heavy dimethylation reagent. One issue which can arise with this technique is the ‘deuterium effect’, where the different number of deutriums between the light, medium and heavy labels can result in shifted retention times for the peptide across the channels [1]. An example of this retention time shift can be seen in Figure 1, where the XIC for the light and medium channels are clearly shifted to the right of the heavy channel.
Click to view full size image
Figure 1: XICs for a light (top), medium (middle) and heavy (bottom) labelled peptide showing the retention time shift across the channels caused by the deuterium effect.
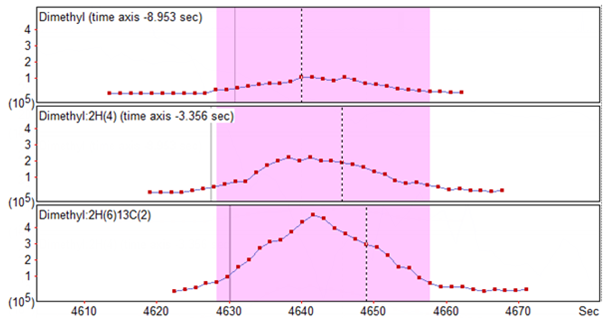
Fortunately, Mascot Distiller can account for this shift in retention time. To enable this, you need to edit the quantitation settings and ensure that the ‘Allow elution time shift’ checkbox is checked, and that you’ve entered a sensible ‘Elution time delta’ to account for the shift between channels – this acts as a ceiling on the retention time shift used to try and align the XICs correctly. Figure 2 below shows the XICs for the same peptide as in Figure 1 where we’ve allowed Distiller to try and align the XICs with a elution time delta of up to 20 seconds:
Click to view full size image
Figure 2: The same peptide as in Figure 1, with the XICs realigned to allow for the retention time shift across the different channels. Distiller has aligned the light and medium signals to the heavy, with a noticeable time shift in both.
Diethylation
In a recent paper published in the Journal of Proteome Research, Jung et al. presented a 3-plex method based on peptide diethylation [1]. This method has many of the same advantages as Dimethylation, whilst avoiding the Deuterium effect on retention time. Mascot doesn’t ship with this new method of course, but it is very easy to add.
Configuration must be done on the Mascot server. The ‘light’ diethyl modification is present in Unimod, but the medium and heavy variants are not. Therefore, the first thing you’ll need to do is to add the medium and heavy component modifications to your Mascot server, via the configuration editor. Make copies of the Diethyl modification and edit the chemical formulae to “13C(2) C(2) H(8)” for the medium modification and “13C(4) H(8)” for the heavy modification.
Once you’ve added the modifications, you can define the quantitation method. In the Mascot configuration editor, add a new quantitation method ‘Diethylation [MD]’
- Set the method to precursor
- Create the “light” component, with a modification group containing the Unimod Diethyl
- Create the “medium” component, with a modification group containing medium Diethyl
- Create the “heavy” component, with a modification group containing heavy Diethyl
- All mod groups should be set to ‘exclusive’
- Add two report ratios, M/L with a numerator of medium, and H/L with a numerator of heavy. Both should be defined with a denominator of light
- Integration and quality settings are also important – see Distiller help (Quantitation Toolbox -> Method Configuration – Precursor) for further details
Comparison
To compare Diethylation quantitation in Mascot Distiller with the Dimethylation method, we took the 1/2/4 Deithylated and Dimethylated raw files from PRIDE PXD012117, where the authors had mixed light, medium and heavy labelled proteins in a ratio of 1-2-4.
The raw files were processed in Mascot Distiller as two separate independent multifile projects, one for the Diethylated samples, and one for the Dimethylated samples. The generated peaklists were then searched against SwissProt with a Human taxonomy filter and the automatic decoy search selected. Other search settings were taken from Jung et al. Once the searches had been imported into Mascot Distiller, the significance thresholds were adjusted to give a 1% PSM FDR, and the data quantitated using the relevant quantitation method. In all cases, the ratios calculated were Medium/Light and Heavy/Light, with expected ratios of 2 and 4 respectively. In addition, to look at the Deutrium effect, the Dimethylated datasets were quantitated twice, once without allowing elution time alignment, and once allowing a time shift of up to 20 seconds. The following quality thresholds were then applied to all the datasets:
- Correlation threshold: 0.7
- Std. Error Threshold: 0.1
- Fraction Threshold: 0.5
| Medium/Light | N | Heavy/Light | N | |
|---|---|---|---|---|
| Expected ratio | 2 | 4 | ||
| Dimethylation(no timeshift) | 1.99 | 444 | 19.32 | 411 |
| Dimethylation(allow timeshift) | 1.95 | 1806 | 3.97 | 1970 |
| Diethylation | 2.05 | 2392 | 4.01 | 2517 |
From Table 1, we can see that all three methods have returned a median M/L value of close to the expected ratio of 2. However, the quality thresholds have removed a very large number of peptides for Dimethylation when no timeshift was allowed. The median heavy/light ratio for Deimethylation without allowing a timeshift is nearly 20, instead of the expected value of 4, whilst removing even more peptide quantitation matches. This is because the light and heavy XICs are badly misaligned, resulting in very low light component signal being used in the calculation. With elution timeshift enabled, Dimethylation performs well, the median ratios now being very slightly below the expected values.
Using Diethylation as a quantitation method compares very favourably with Dimethylation + timeshift. The median ratio values are very close to the expected ratios, and more peptides have passed the quality thresholds when compared with Dimethylation.
References
- Jung J, Jeong K, Choi Y, Kim SA, Kim H, Lee JW, Kim VN, Kim KP, Kim JS. J. Proteome Res. 2019 Mar 1;18(3):1078-1087
Keywords: deuterium, diethylation, dimethylation, Mascot Distiller, quantitation