Spectral library search
One or more spectral libraries can be searched, either alone or in combination with Fasta databases. The search engine for spectral library files is MS PepSearch from the NIST Mass Spectrometry Data center. Whenever a search includes a spectral library, the results are presented using the Protein Family Summary. Protein inference for library matches is accomplished by assigning a reference Fasta database to each library as part of the library configuration. Each match to a peptide sequence in the library is mapped to all the protein entries in the reference Fasta that contain that same sequence, which enables rigorous parsimony-based protein inference to be performed.
This example shows the results a simple search of a single spectral library file. The MS PepSearch scores are displayed and assigned a default significance threshold of 300. The library was NIST_S.cerevesiae_IonTrap and the reference database was SwissProt with a taxonomy filter of S. Cerevisiae.
The proteins are grouped according to shared peptides. Expand hit 2 and you can see how G3P3_YEAST and G3P2_YEAST are very similar, while G3P1_YEAST has more differences, and this is illustrated by the accompanying dendrogram. For more details on the features of the protein family summary, see the help page.
You will also notice that there are matches to peptides modified with Carbamidomethyl and Oxidation, even though no modifications were specified for the search, because these modifications were present in the library entry. Likewise, there are some matches to non-tryptic peptides (e.g. N.CLAPLAK.V) even though the enzyme in the search form was trypsin. Only a few search parameters are relevant to a library search, the most important being precursor and fragment mass tolerances.
Compare this library-only search with a search of the same library plus SwissProt. As with a conventional database search, all of the search parameters apply to the Fasta part of the search. At first glance, the results look similar, which indeed they are. As in any multi-database search, accessions are prefixed by an index. In this particular search, the index is 1 for the library and 2 for the Fasta. If you expand family 2, you’ll see that there are many additional matches in the library plus Fasta search. In a few cases, these are peptides that are in the Fasta but not in the library, for example K.ELDTAQK.H. In most cases, the additional matches are peptides that are in both and gave significant matches to the Fasta but not to the library. (The default mode for the report is to show just significant matches.) If you expand all the matches for a query using the twisty in the rank column, you’ll usually see matches to both the Fasta and the library. Sometimes the library match is the stronger, sometimes the Fasta.
Presenting an integrated view of Fasta and library matches, with meaningful protein inference, requires expect values to be assigned to the library matches, as described in the Scoring and Statistics section.
Finally, you can create libraries from your collection of Mascot result files. This can be a one-time process or you can schedule the library to be updated from new result files at regular intervals. For any particular library, filters can be defined, so that it only contains iTRAQ peptides or phosphopeptides or human proteins or whatever is required.
Search parameters
Most search parameters – modifications, enzyme, missed cleavages, taxonomy, and instrument – simply don’t apply to a library search. All that matters is how well the experimental spectrum matches the one in the library. The main exceptions are the precursor and fragment mass tolerances.
The score for the match will have some dependence on the fragment tolerance specified in the search form. This is combined, by root sum of squares, with the estimated average fragment tolerance for the library entries, specified during library configuration. The default library tolerance is quite wide, 0.5 Da / 500 ppm, because the entries may come from any type of instrument, and having a tolerance that is too wide is much better than one that is too narrow. If you are creating a library from data acquired on a specific instrument, capable of high MS/MS accuracy, you may be able to use a much tighter fragment tolerance.
The precursor tolerance specified in the search form is used to filter matches by limiting the difference allowed between the experimental and calculated peptide masses. Note that the accuracy of the instrument used to acquire the library entry was taken into consideration when the entry was added to the library, in that the identity of each entry comes from database searching, but it is not a factor during a search of the library. For example, imagine a library entry created using data from a low accuracy instrument. Maybe the original database match is GDLGIEIPAPEVLAVQK and the original experimental precursor is 874.67, 2+ corresponding to Mr = 1747.32. The calculated peptide Mr is 1747.9720, a difference of 0.65 Da, but the match is a very strong one, so we feel confident enough to add this spectrum to our library. If we then search this library with data from an instrument capable of low ppm accuracy, and we get a match to this entry, we accept or reject the match according to whether the experimental peptide mass is within (say) 5 ppm of the calculated mass for GDLGIEIPAPEVLAVQK. The identity of the library entry may or may not be correct, but the original mass error of 0.65 Da is not relevant.
Mass values in a library search must always be monoisotopic.
Peptide charge is not used to filter matches. Mainly, because it can easily be wrong and one of the advantages of a library search is that this doesn’t matter. You may have a spectrum with a supposed charge of 2+ get a strong match to a library entry with a charge of 3+. However, note that some peak picking utilities handle ambiguous charge by outputting duplicate spectra, each one taking a different precursor charge from a specified range, in the hope that one will match. In a spectral library search, where charge doesn’t matter, all will match, inflating the PSM count.
When a search includes a library, you cannot check decoy or error tolerant. We hope to introduce library target-decoy in a future release. Error tolerant mode doesn’t apply to library entries, since we don’t specify modifications or enzyme specificity.
In summary, a complete list of the search parameters that apply to a library search is:
| Name | Description |
|---|---|
| COM | Search title |
| CUTOUT | Precursor removal |
| DB | Database |
| FORMAT | MS/MS data file |
| ITOL | Fragment ion tol. |
| ITOLU | Units for ITOL |
| LOCUS | Hierarchical scan range identifier | PEPMASS | Precursor m/z |
| RAWFILE | Raw file identifier |
| RAWSCANS | Native scan range identifiers |
| REPORT | Maximum hits |
| RTINSECONDS | Retention time or range (in seconds) |
| SCANS | Scan number or range |
| SEARCH | Type of search (must be MIS) |
| TITLE | Query title |
| TOL | Peptide mass tol. |
| TOLU | Units for TOL |
| USER00 to USER12 | Uncommitted parameters |
| USEREMAIL | User email |
| USERNAME | User name |
Reports
The report format for a library search is always the protein family summary. If the search is library only, the format controls are reduced to Significance threshold with a default library score of 300, Max. number of families, a checkbox for Display non-sig. matches, and Dendrograms cut at.
When a mixture of library and Fasta files are searched, the results are integrated into a single report. There is an additional format control for Report mode with choices of Integrated, Only Fasta, and Only library. The latter two choices allow you to see the results from just one class of file, in case this is useful. There may be additional controls that are only relevant to the Fasta results, such as Preferred taxonomy. Percolator is not available for a report that includes library matches.
Auto-decoy is not available when a library file is searched, but the Sensitivity section in the report header can be opened to see counts for PSMs and distinct sequences for the complete set of results. You can choose to display counts for all matches, or just those from the Fasta or library files. For Fasta matches, you can choose between the identity and homology thresholds.
The tables of peptide matches in the body of the report have an additional column headed source which shows AA for amino acid Fasta, NA for nucleic acid Fasta, XA when both AA and NA Fasta files have been searched and the match is found in entries from both, or SL for library. Note that library and Fasta matches are always shown as different ranks and it is common for the same sequence to be found in both. Because Fasta scores and library scores are on very different scales, the ranking is by expect value, as explained under Scoring and Statistics.
In the example of a simple library search, all of the accessions visible in the summary report are from the SwissProt reference database. Click on an accession link to load a protein view report, and you’ll see that the SwissProt entry forms the basis of the report.
Click on a query number link to load a peptide view report. Towards the top of the report, the observed and library spectra are shown as mirror images, illustrating the quality of the match. To zoom in, drag out the desired range in the middle of the graphic, between the two horizontal axes. Peaks that match are highlighted (blue by default). The annotations for the original library entry are reproduced towards the bottom of the report.
Reference Database
MSP accessions are those present in the original, MSP format spectral library file. If an entry has no MSP accession, we assign the peptide sequence as the MSP accession. MSP accessions are not useful for protein inference:
- Peptide sequence as accession means that duplicate matches group together, but this doesn’t help with protein inference
- MSP accessions may be meaningless
- Usually, there is only a single MSP accession, so protein inference will be very limited for shared peptides
- MSP accessions may be from different sources, e.g. some from UniProt, some from NCBI
To make protein inference feasible, we require the user to specify a reference Fasta file for each library. The reference Fasta must contain amino acid sequences – nucleic acid is not allowed. Each library entry is assigned to all the accessions from the reference file that contain the peptide sequence, ignoring enzyme specificity. This means that, in favourable cases, protein inference will be just as good as if the matches had been found in a search of the references Fasta file.
There are, of course, limitations:
- Reference file may be badly chosen, e.g. poor coverage or even wrong species
- Some peptides will fail to map because the Fasta is incomplete or the peptide is an unusual variant
When an entry has no reference accession, we use the original MSP accession(s). When the reference file is well chosen, as in the example, few or no MSP accessions will be visible in the summary report.
When both Fasta and library files are searched, in an integrated report, Protein inference will use accessions from the Fasta files in preference to accessions from the library. A library accession will only be used if a PSM cannot be mapped to any Fasta entry. This means that we only ever see a library accession in an integrated report when it is the anchor protein for a family member. If it was a same-set or sub-set or intersection protein, the library accession would be discarded.
Scoring and Statistics
Scores from NIST MS PepSearch range from 0 for no match to 999 for a perfect match. When the results contain fewer than 20 significant matches from Fasta file, possibly because only library files were searched, the score is assigned a fixed significance threshold of 300. This is a near arbitrary value intended to filter out random matches. Expect values are calculated in a similarly arbitrary way using the following expression:
E(s) = 0.05 * (10 ^ ((300 – s) / 100))
Where E(s) is the expect value for library score s. This gives an expect value range from 50 for score 0 to 5E-9 for a score of 999. This is done to ensure that reports and exports containing library results can be handled using the same code as Fasta results. There is no suggestion that the significance threshold or expect values are in any way accurate or statistically meaningful.
For an integrated report to work, the expect values for the library and Fasta matches need to be on a similar scale. This is achieved by scaling the set of queries where the library and Fasta return the same match and the Mascot score is significant. Expect values are assigned to the library scores so that the distributions have the same mean and variance for both library and Fasta.
You can see this behaviour by opening the score distribution section of the report header. For the example search of just a library, the threshold is 300 and, in the body of the report, a library score of 670 is assigned an expect value 1.0e-05. In the integrated report for a search of library and Fasta, the threshold is adjusted to 493 and a library score of 670 is assigned an expect value 8.6e-05.
MS PepSearch
To perform a library search, NIST MS PepSearch version 0.93 is executed with the following arguments:
MSPepSearch.exe m a P /ZPPM 100 /M 0.509902 /LIB [path to library] /INP [path to MGF peak list] /OUTTAB [path to output file] /HITS 10 /MinMF 0 /NumCompared /OutPrecursorMz /OutDeltaPrecursorMz /OutSpecNum
| Argument | Description |
|---|---|
| m | pre-search requires precursor m/z to be within tolerance specified by /Z |
| a | alternative peak matching |
| P | use peak annotations and peak weighting |
| /ZPPM 100 | precursor tolerance (varies according to search parameters) |
| /M 0.509902 | fragment tolerance (varies according to library tolerance and search parameters) |
| /HITS 10 | max. number of output hits |
| /MinMF 0 | min. match factor to output |
| /NumCompared | output number of compared spectra |
| /OutPrecursorMz | output precursor m/z |
| /OutDeltaPrecursorMz | output hit-unknown precursor m/z delta |
| /OutSpecNum | output spectrum number |
To create a searchable set of binary files, a command line version of Lib2NIST is executed with the following arguments
lib2nistcl.exe -log9 [path to log file] -msp2peplib [path to MSP file] [path to output directory]
Library Configuration
Mascot Server (2.6 and later) supports three file formats for spectral libraries:
The sptxt format is a variation on MSP. The main functional difference between MSP and MGF is in the level of annotation. The X!Hunter MGF format is minimalistic and has only a few match-level items of metadata (sequence, modifications, precursor mass, charge). The peak list is unannotated. In contrast, MSP files usually have a wealth of annotation down to individual peak level.
Adding and updating spectral library files is handled by Database Manager, just as for Fasta files. A number of libraries from NIST and PRIDE are ‘pre-configured’. All you have to do in Database Manager is choose Enable predefined definition from the Library menu, then Enable the library of interest. The Wizard will ask you to confirm the location for the library files and the identity of the reference database. This defaults to SwissProt with a taxonomy appropriate to the library. You can select any locally available Fasta file, but we advise against choosing a very large database, such as NCBIprot, even with a restricted taxonomy, because the huge number of proteins mapping to each peptide sequence will make compression, searching, and reporting very much slower than with a less redundant file, such as SwissProt or a UniProt complete proteome.
Choose Create new to create a custom library definition. After choosing a name for the database, you specify the source. This can be an existing library file that is copied or downloaded or the library can be created from your collection of Mascot search results.
For a custom definition, once the source has been specified and a library file either created, downloaded, or copied, the wizard has three further steps:
- Choose accession and description parse rules for the library protein accessions. These are not required if the library does not contain protein accessions, as is the case with most Pride libraries.
- Supply estimates (Da and ppm) for the precursor and fragment mass errors of the library entries. If unsure, too wide is better than too narrow
- Specify a reference database and, if relevant, taxonomy. Choosing a suitable reference is critical to getting good protein inference.
Some spectral library files contain non-standard modification names, which creates problems in the result reports. A file called library_mod_aliases in the Mascot config directory contains mappings between names found in spectral library files and Unimod / PSI modification names.
Syntax is ‘name used in library file’ = ‘Unimod / PSI name’. Lines that start with # are comment lines. Any modification in any spectral library with the name shown on the left will be replaced with the name on the right before being saved as the .nist_format file, which is used by MSPepSearch.
Creating a Library from Search Results
If you choose to create your own library from search results, you must supply mass error estimates and reference database details, as above. You then specify at least one ‘peptide match filter’, which must be a score or expect value threshold, typically expect < 0.01. You can have many filters, and each individual filter is in a filter group. To add more filters to the group, use the OR button. To add more groups, use the AND button. The peptide match must pass all filter groups to be accepted, but within each group, only one filter needs to succeed.
To give an example that illustrates the flexibility of this arrangement, here is a set of filters that will select searches run by either Romulus or Remus and matches that have expect values less than 0.01 and are phosphopeptides and are from a human protein.
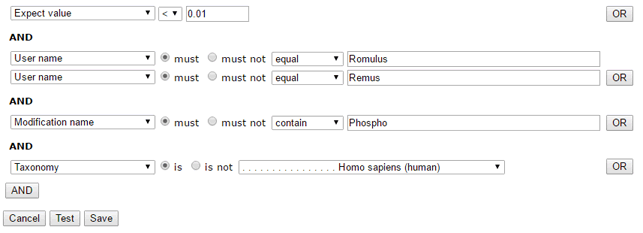
The next step is to import search results filtered by date range and an optional filepath wildcard. Later on, you can schedule a recurring update task to import matches from new Fasta search results.
If you have enabled tidy_data.pl in the Cron section of mascot.dat, to compress old result files and delete cache files, be careful when scheduling recurring tasks to update libraries from new Fasta search results. One process may be trying to compress result files while the other is trying to decompress them, and there is the possibility of conflict. Best to schedule these processes for different days of the week.
References and Acknowledgements
We are grateful to Stephen Stein, Dmitrii Tchekhovskoi, Yuri Mirokhin and the team at NIST for developing and maintaining the MSPepSearch software. We also thank Paul Rudnick of Spectragen Informatics for custom development and consultancy. Information on the NIST software and libraries can be found here. The primary publication for MSPepSearch is:
These two publications describe the PRIDE spectral libraries:
- Griss, J., Foster, J. M., Hermjakob, H., and Vizcaíno, J. A., PRIDE Cluster: building a consensus of proteomics data, Nature Methods 10, 95–96 (2013)
- Griss, J., et al., Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets, Nature Methods 13, 651–656 (2016)
Sources of spectral libraries:
