Exporting search results: tips and tricks
The Mascot Server export utility allows search results to be exported in a wide range of formats, together with the native result file and the MGF peak list. Exporting the result file can be useful if you need it for a third party application and don’t have file share access to the Mascot Server. Exporting the MGF might be useful if you lost the original. Note that the exported MGF will not be perfectly identical to the original. The sort order will be different, although this shouldn’t matter, the intensity and mass values may have been rounded according to the mascot.dat settings for MassDecimalPlaces, IonsDecimalPlaces, and IntensitySigFigs, and some peaks may have been merged due to re-centroiding, as discussed in this blog article.
The export utility can be used interactively, by following the appropriate link in a result report. It can also be executed automatically by Mascot Daemon, after each search in a task. You can export a batch of existing result files by creating a Windows batch file or Linux shell script. The help page for exporting explains how to do this, and is essential reading. If you aren’t familiar with such scripts, some aspects may be confusing. The script must be saved in the Mascot cgi directory on the Mascot Server; it cannot be executed from an arbitrary directory or from a remote PC. The command-line arguments for a given output can be obtained by entering the required settings in the browser form then choosing "Show command line arguments". The output must be piped to a file by adding a > symbol followed by the path to the output file. This can be an absolute or relative path to any location for which you have write access.
Mascot reports offer many formatting choices, such as the number of protein hits and a score or expect value cut-off to filter out weak peptide matches. In most cases, if you load the export form from the search result page, these settings are carried through, and you’ll get exactly the same numbers in both. If you perform the export from Daemon or a script, and load the report independently, this may not be the case. There is a table near the top of the help page that shows key settings equivalent to the main result reports.
We are often asked about exporting site analysis data – the information displayed in a Peptide View report when alternative arrangements of variable modifications are possible. If you look at a CSV export, site analysis is not included in the main peptide and protein ‘table’ because it needs to be seen in the context of the other possible matches. A value of 60% would be interpreted very differently according to whether the next best arrangement is 40% or 1%. To obtain site analysis values in the CSV and XML export formats, you have to check Query Level Information and Raw peptide match data. It is then output as a separate section, after the main protein and peptide table.
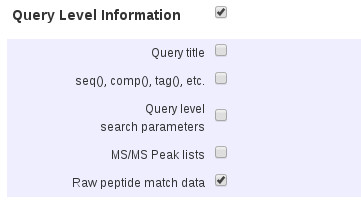
There is a checkbox for emPAI quantitation under Protein Hit Information. If you want to export MS2 quantitation results (reporter or multiplex) check the boxes under both Protein Hit Information and Peptide Hit Information. In all cases, in the CSV export, the protein quantitation values can be found in the row for the final peptide match for each protein, to the right of the main table, without column headers.
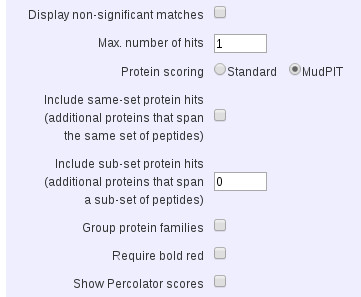
If you want a peptide-centric CSV export, showing just the top match for each query, without protein inference, you have to do a little bit of re-formatting. Set Max. number of hits to 1, Include sub-set protein hits to 0, and uncheck Include same-set protein hits and Group protein families. Under Peptide Hit Information, check Unassigned queries. When you open the CSV in Excel, you’ll see a main table for one protein followed by the unassigned list. Select the main table and sort by pep_rank. If any rows have values of pep_rank greater than 1, delete them. Also, delete the three empty rows between the main table and the unassigned list. Select both the main table and the unassigned list and sort by pep_query.
There are complete blog articles dealing with mzIdentML and mzTab. Some complications associated with exporting retention time are discussed in the Retention time blog article. Finally, if you have been puzzling over how to export library search results, please give us a little more time. We weren’t able to squeeze this into the 2.6.0 release, but it is slated for 2.6.1.
Keywords: export, MGF, site analysis