Solving a puzzle with Mascot Distiller de novo
Have you tried either of the MS/MS interpretation challenges organised by EUPA and LBMSDG? Maybe you are expert enough to read off the peptide sequence, directly. If so, you can stop reading right here!
If your interpretation skills and mental arithmetic are a bit rusty, we thought it might be useful to walk through how Mascot Distiller plus a bit of guesswork can be used to find the solution to the second challenge. If you don’t already have Distiller, you can get a free, 30 day trial.
The data are presented as a PDF of a labelled spectrum. The most tedious part of the exercise is getting the mass values into a machine readable peak list. Depending on how you view the PDF, you may be able to type CTRL+A and CTRL+C to select all the text and copy it to the clipboard. If this works, paste the text into a spreadsheet and clean it up by deleting the axis labels and titles. If this doesn’t work for you, you’ll just have to type the mass values in.
You also need an intensity value for each row. Distiller de novo doesn’t care what the intensity values are, so you can add a second column with the same number in every row. Or, if you prefer to see something more realistic, you can estimate peak heights – they don’t need to be accurate. You might also wish to sort the values by mass, but this is not essential. Save the data as tab delimted text. If you’re in a hurry, you can download the peak list file.
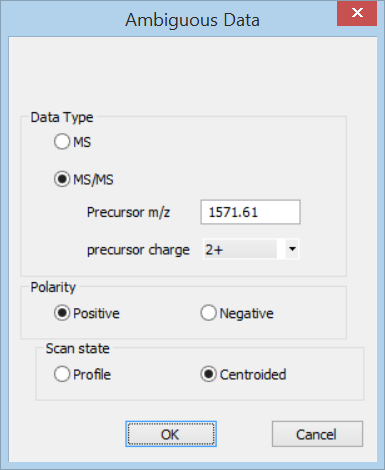
In Distiller, choose New Project, Text from the File menu. Select the peak list and enter the precursor information in the Ambiguous data dialog.
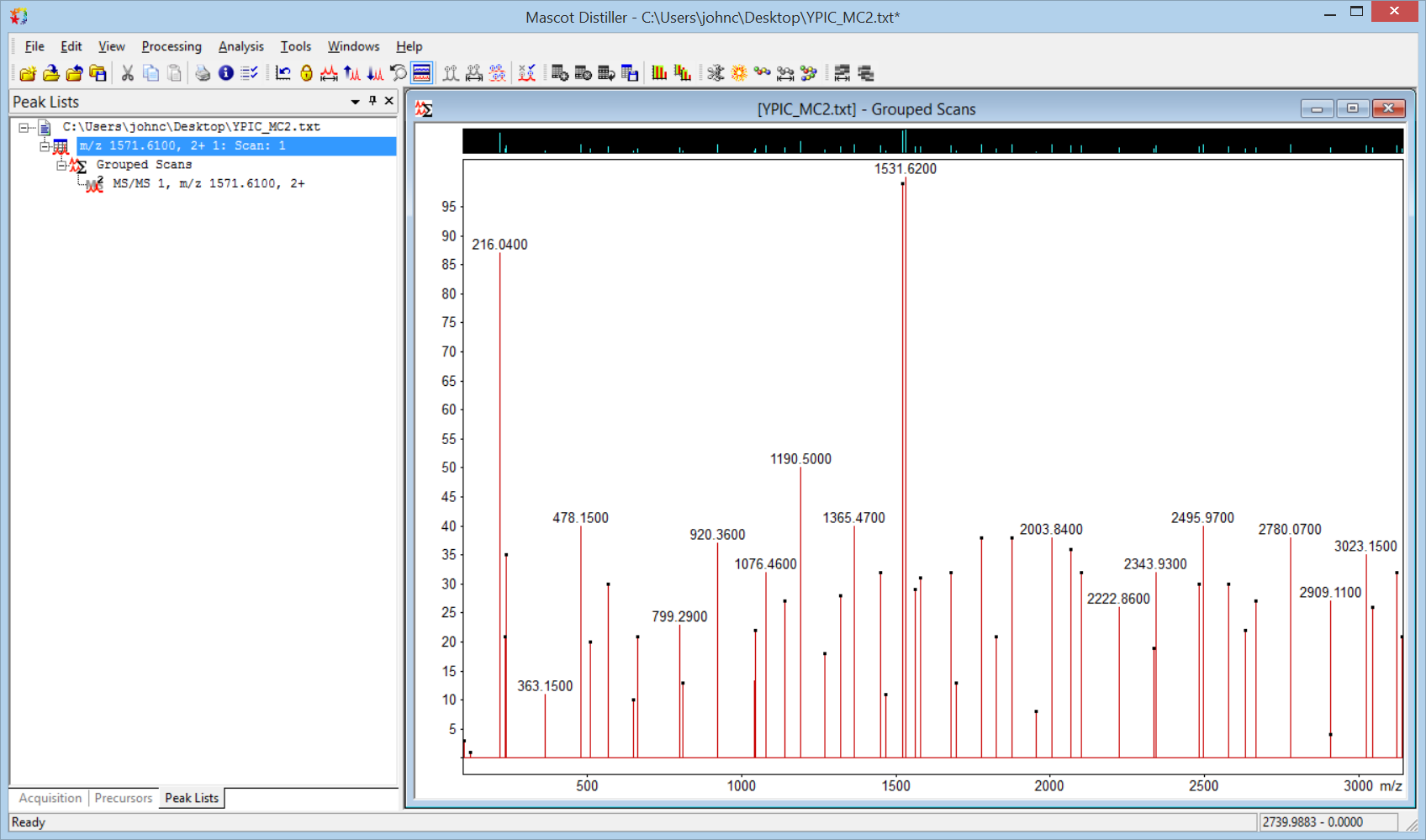
You should now have the spectrum in Distiller, ready for peak picking. The default processing options for text need a couple of tweaks. On the MS/MS processing tab, clear Use precursor charge as maximum and set Maximum charge to 1. On the MS peak picking tab, under General, clear Apply baseline correction and change Fit method to Single peak. Under Peak profile, set all three width values to 0.02. Choose Process scan from the Processing menu or the toolbar. You should now have something similar to this:
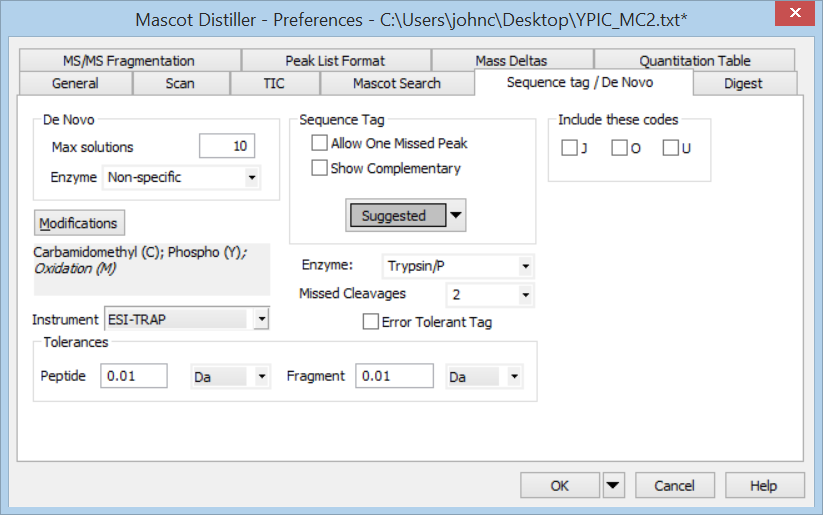
From the Tools menu, choose Preferences, Sequence tag / De Novo. Since we cannot assume this is a tryptic peptide, set Enzyme to Specific and non-specific. Change Peptide tolerance to 5 Da, because we’ve been told the peptide is modified in unknown ways, so we don’t want to constrain solutions to the precursor mass at this stage. Change Fragment tolerance to 0.1 Da, which is a guess based on the labels all having two decimal places. Choose OK, then Denovo search from the Analysis menu.
Several similar solutions will be displayed, the top three being
PHqDS[YA|SF|PH|MC][WG|SR|NE|Dq]PEPTPMEENR[EE]PSDE[SK]
PHqDS[YA|SF|PH|MC][WG|SR|NE|Dq]PEPTiDEENR[EE]PSDE[SK]
PHqDS[YA|SF|PH|MC][WG|SR|NE|Dq]PEPT[VE|PM|NN|Di]EEN[YN]HPSDE[SK]
If this was a genuine peptide, with three unknown modifications, it would be a struggle to reach an unambiguous solution. Since this is a competition, we might hope that there are some clues to guide us along. If you stare at these three solutions for a while, you may notice the word PEPTIDE in the middle of the second solution. This is unlikely to be a coincidence. It’s hard to make out any English words to the right of the solutions, but they all begin PHqDS[PH][Dq]. If we substitute qD or Dq with the letter O, we now have PHOSPHOPEPTIDE
The instructions stated that the sequence included a non-canonical letter, so something like this is to be expected. The mass of qD is 243.08. If you look at the delta between this and the masses of each of the amino acid residues, the value which jumps out is 80 for Tyrosine. So, the hypothesis is that one of the modified residues is pY and this is represented in the sequence by O.
In Distiller Preferences, Sequence tag / De Novo tab, enter Phospho (Y) as a variable modification. When de novo is repeated, the highest scoring solution is
PHYSPH[WG|SR|NE|Dq]PEPTPMEENRi[GC]HFENT
For some reason, the algorithm is still choosing Dq rather than Y for the second O of PHOSPHO, and PEPTIDE is going slightly adrift, but we can now see that the right hand side is not very different from ENRICHMENT. The deltas would be two of the most commonly encountered modifications: GC is isobaric with carbamidomethyl Cys while F is nearly isobaric with oxidised Met. Add Carbamidomethyl (C) as a fixed modification and Oxidation (M) as variable, and the highest scoring solution becomes
PHqDSPHYPEPT[VE|PM|NN|Di]EENRiCHFENT
The algorithm is reluctant to display the exact set of alternatives we would like to see, but the calculated mass exactly fits the precursor mass and we have excellent, end-to-end sequence ion coverage. You can confirm the solution by entering the deduced sequence in the Distiller fragment ion calculator or by tightening up the tolerances and making Phospho (Y) a fixed modification.
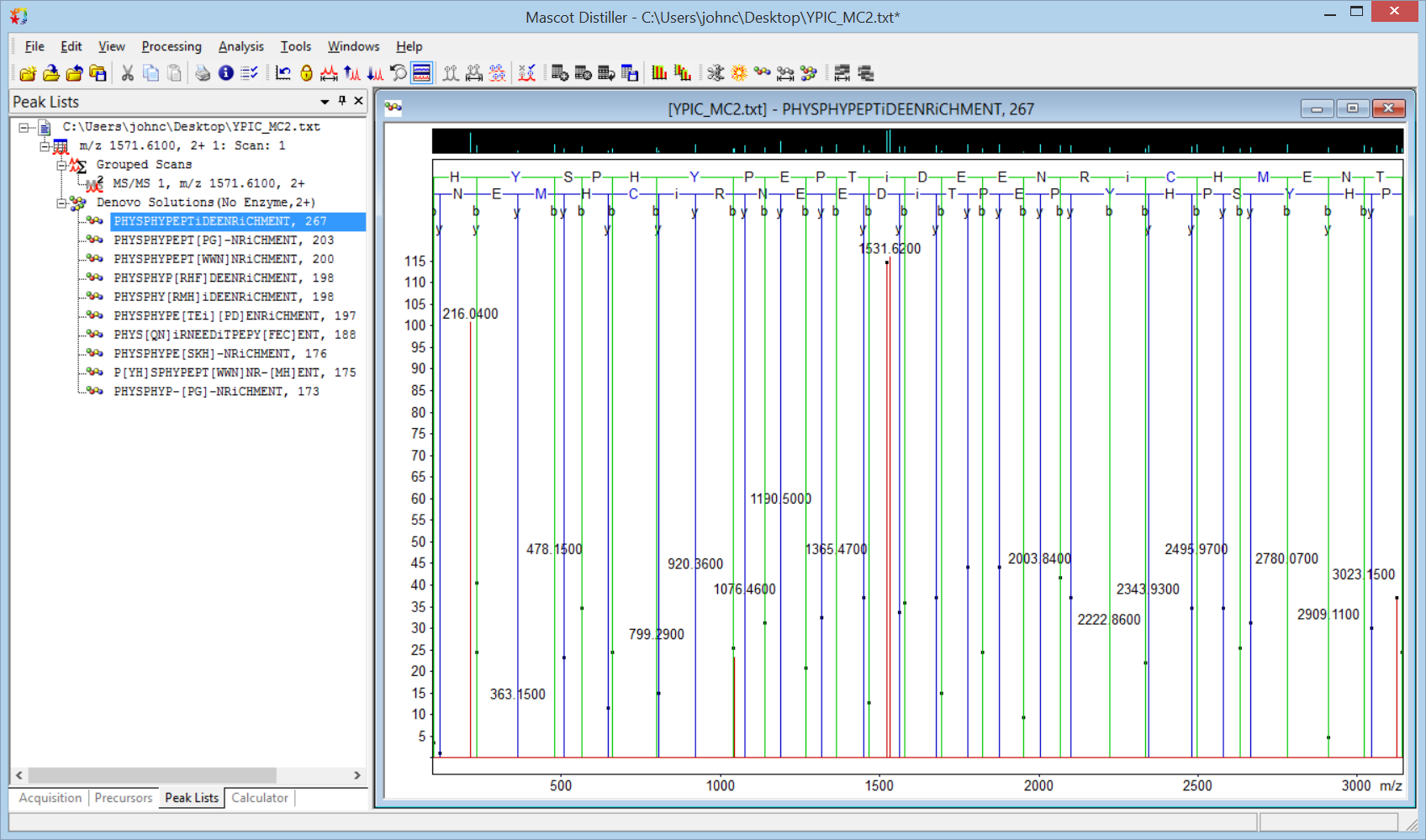
Keywords: de novo, interpretation, Mascot Distiller, tutorial
Crediting the MS/MS interpretation challenges to EUPA and LBMSDG was inaccurate. Should have read EUPA/YPIC and LPDG.