What are you inferring?
Benchmarking protein inference is notoriously difficult. Artificial samples of known content tend to be too simple while real samples lack ground truth. An interesting approach was adopted for the ABRF iPRG 2016 study, and has been the subject of a publication from The et al.
A collection of human Protein Epitope Signature Tags (PrESTs) were expressed in E. coli and 191 overlapping pairs selected so that each pair would have some unique and some shared tryptic peptides. The pairs were divided into two pools, A and B, and a third pool was created by combining A and B. Samples from the pools were spiked into an E. coli cell lysate and analysed in triplicate. All files can be found at Pride project PXD008425.
We used Mascot Distiller 2.7 for peak picking and Mascot Server 2.6 for searches. An error tolerant search showed the peptides to be seriously over-alkylated. For example, the modification counts for the pool A files, out of a total of 9739 significant PSMs, were:
Modification Site Count Carbamidomethyl C 4062 Carbamidomethyl N-term 3925 Carbamidomethyl M 2348
Hence, search parameters were set as follows:
Enzyme : Trypsin/P Fixed modifications : Carbamidomethyl (C) Variable modifications : Carbamidomethyl (M), Carbamidomethyl (N-term) Mass values : Monoisotopic Peptide mass tolerance : ± 10 ppm Fragment mass tolerance : ± 10 ppm Max missed cleavages : 2 Instrument type : ESI-TRAP
Three Fasta files were provided:
- prest_1000_random (1,000 sequences; 66,666 residues)
- prest_pool_a (192 sequences; 20,173 residues)
- prest_pool_b (191 sequences; 13,451 residues)
prest_1000_random is a collection of decoy PrEST sequences that can be used to estimate protein FDR.
| Search | Sample | Database | Peptide FDR | PrEST pool A | PrEST pool B | PrEST random | E. coli |
|---|---|---|---|---|---|---|---|
| 5148 | A+B | PrEST | 0.7% | 174 | 181 | 0 | N/A |
| 5149 | A+B | PrEST + E. coli | 1.3% | 174 | 181 | 0 | 538 |
| 5142 | A | PrEST | 0.8% | 179 | 7 | 0 | N/A |
| 5144 | A | PrEST + E. coli | 1.2% | 179 | 7 | 0 | 621 |
| 5143 | B | PrEST | 0.8% | 6 | 185 | 0 | N/A |
| 5146 | B | PrEST + E. coli | 1.0% | 5 | 185 | 0 | 546 |
For the Mascot search of the A+B pool against the combination of the three PrEST databases, at a distinct peptide sequence FDR of 0.7% and applying a two peptide rule, the counts of proteins were as shown in the first row of the table (search 5148). For pool A, there are 192 Fasta entries but only 174 proteins in the report. In 15 cases, this is because the protein has no unique PSM and becomes a sub-set protein, which is relegated from the minimal list. For 3 entries, there are no matches, not even peptide molecular mass matches. For pool B, the 10 "missing" proteins are 8 subsets, 1 sameset, and 1 entry with no significant PSMs.
The three PrEST databases, a total of 100k residues, represent a tiny search space compared with a typical proteome. To create a more realistic search space, we included the Uniprot proteome for the host cells, E. coli (strain B / BL21-DE3), which increased the size by more than an order of magnitude, to 1400k residues. Reassuringly, the PrEST counts for the A+B pool were identical (second row of the table)
For the search of pool A proteins, the count of 7 proteins from pool B seems, at first sight, a little high. On closer examination, there appears to be some cross-contamination. For example, the raw files for pool A contain 4 high scoring PSMs to TLLDYWQALENSRGEDCPPV, a peptide that is only present in the pool B Fasta. It is very hard to believe that this is a false positive; compare Scan 33715 from mixtureArep1.raw (false positive?) with Scan 34730 from mixtureBrep3.raw (true positive).
Some of these matches must be sample injection carry over, because there are also high scoring matches to PrEST peptides in the blank runs, particularly the second replicate. It would seem that getting zero counts for matches to prest_1000_random is a safer check on protein FDR than counts to pool A sequences from the pool B sample or vice versa. In terms of protein inference, the Mascot protein family summary report seems as accurate as the data will allow.
While the sample is more realistic than many, the database is artificial, in that it contains perfect representations of all the target sequences. This is not the case for most real-life searches, which use a public database. Due to natural variants, the sequence of a particular protein in the database will often differ from the sequence of the protein in the sample. If we get matches to two similar sequences, the truth may be that the analyte is partly represented by one database entry and partly represented by the other. In such cases, it is misleading to report that only one is present or that both are present.
In their paper, The et al. state that "inference procedures excluding shared peptides provide more accurate estimates of errors compared to methods that include information from shared peptides". This is very much a consequence of using an artificial database, and should not be taken as gospel. Consider a more realistic, non-identical database containing two homologous protein sequences, maybe 90% identical. Whether we get matches to one or both, unless the coverage is almost complete, excluding shared matches will mean that neither protein appears in the result report, which cannot be acceptable.
You can mask inference problems by selecting a highly non-redundant database, that collapses similar proteins into a single, canonical sequence. This may be fine for a rough survey, but the drawback is that some PSMs are lost because the target peptide sequence didn’t make it into a canonical sequence. For a well characterised organism, the most sensitive and accurate search results come from searching a database such as a Uniprot proteome with isoforms. (You can choose whether or not to include isoform sequence in the downloaded Fasta. For the human reference proteome UP000005640, canonical sequences give a database of 32 MB and 74,416 entries, adding in the isoforms increases this to 47 MB and 96,418 entries.)
If we take the A+B pool and search it against the combined E. coli and human proteomes, we might expect to get 191 human proteins, because the PrEST samples are 191 overlapping pairs of sequences from the same protein. In fact, at 1% peptide FDR and applying the two peptide rule, we get 227 proteins. This is because of the greater complexity of the database, which provides additional permutations of PSMs. For example, in the PrEST databases, we have this pair:
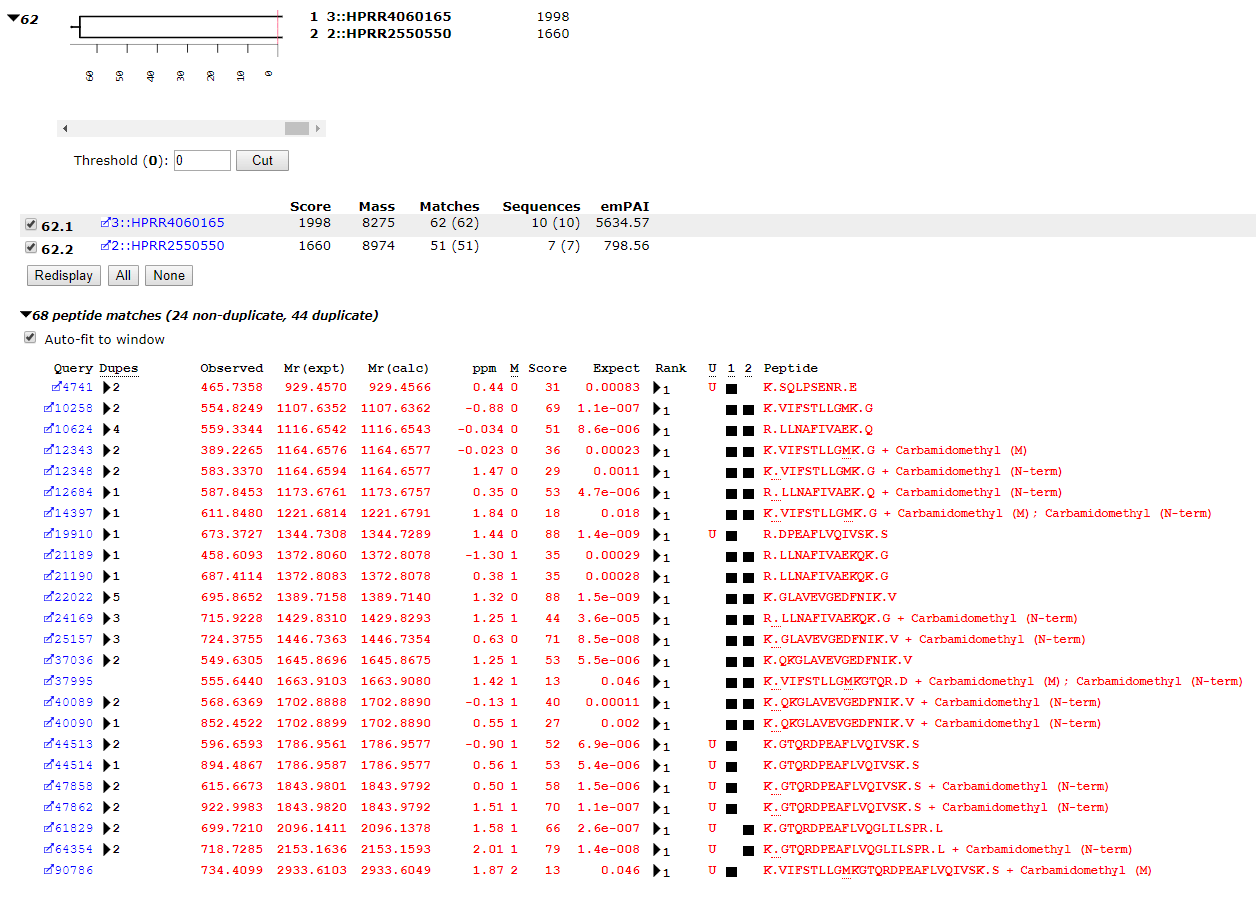
In the human proteome, as seen in the screenshot below, this set of PSMs maps to a pair of isoforms of CENPL_HUMAN, each of which has unique matches. R.DPEAFLVQIVSK.S and K.GTQRDPEAFLVQIVSK.S are only found in isoform 1 while K.GTQRDPEAFLVQGLILSPR.L is only found in isoforms 2 and 3, (in the Mascot report, isoform 3 becomes an intersection protein, because all of the peptide matches can be accounted for by the other two isoforms).
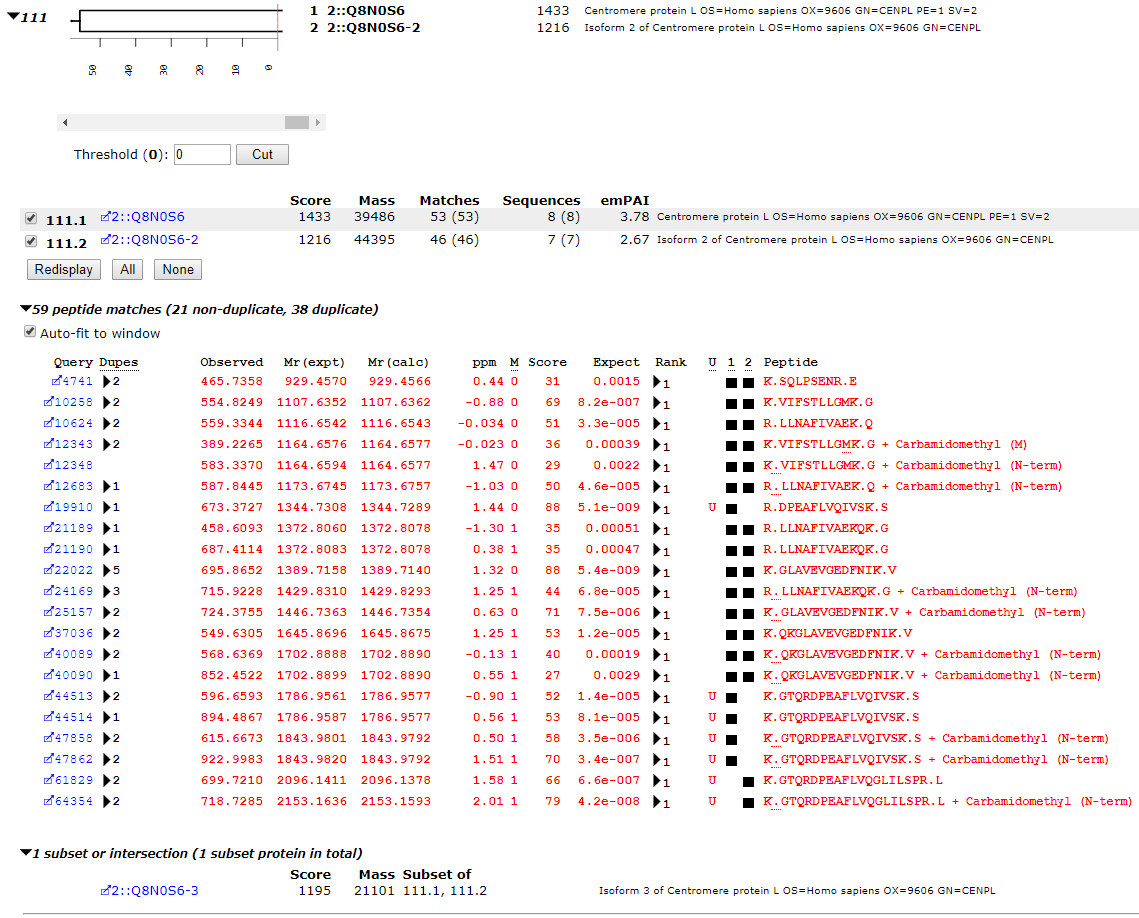
If we searched only the canonical sequences, we would not see the matches to K.GTQRDPEAFLVQGLILSPR.L and assume we had a single protein. If we searched the complete database but excluded shared PSMs, we would discard the evidence that favours isoform 2 over isoform 3. Also, if a two peptide rule was applied, we would only report a single protein, which again, is incorrect. We have ground truth for this sample, and the peptides map to two different splice variants.
Discarding reliable matches or searching an over-simplified database seems short-sighted, at best. There is complexity and ambiguity to real samples and real databases that cannot be reduced to a simple ranking by protein score. Statistics cannot resolve this uncertainty. What matters is presenting the evidence clearly, as in the protein family summary. If a protein such as CENPL_HUMAN is not of interest, we don’t need to waste time on it. If, on the other hand, it is important to our line of research, and there is ambiguity, then proper searching and reporting allows us to probe deeper or design a targeted experiment.
Keywords: benchmark, carbamidomethyl, Fasta, FDR, protein inference, statistics