Automatic target-decoy searching
Many journals impose guidelines for the reporting of database search results, designed to ensure that the data are reliable. The current MCP guidelines require:
"For large scale experiments, the results of any additional statistical analyses that estimate a measure of identification certainty for the dataset, or allow a determination of the false discovery rate, e.g., the results of decoy searches or other computational approaches."
The Human Proteome Project data interpretation guidelines have the same requirement, as do many other journals.
Mascot combines probability-based scoring with an empirical estimate of the false discovery rate. This is implemented by running an automatic target-decoy search.
Automatic decoy search
Mascot always run an automatic target-decoy search. During the search, every time a protein sequence from the target database is tested, a decoy sequence of the same length is automatically generated and tested. The matches and scores for the decoy sequences are recorded separately in the result file. When the search is complete, the numbers of matches and the false discovery rate (FDR) are reported in the result header.
This screenshot shows an example of the decoy statistics for an MS/MS search as displayed in the Protein Family Summary.
Requirements
Automatic target-decoy searching is always enabled for every search type, except:
- Intact crosslink searches – decoy search is not currently supported; see Validating intact crosslinked peptide matches.
- Spectral library searches – decoy search is not currently supported; see spectral library match scoring.
With unsupported search types, Mascot simply runs the search against the target database or spectral library.
A decoy search is run regardless of the size of the search. However, decoy statistics for searches with very few queries, or searches against very small databases, are unreliable, as the sample size is too small.
Turning off automatic target-decoy searching
It is possible to turn off automatic target-decoy searching, but it is rarely necessary.
There are two approaches to how the target and decoy results are integrated: empirical null (EN) and target-decoy competition (TDC). Mascot uses the empirical null approach, where the decoy matches provide an empirical model for the incorrect matches in the target database. The decoy results are used for estimating the FDR and have no other effect on the results from the target database. If you select a target FDR, an appropriate score threshold is set based on the estimated FDR, but no peptide matches from the target database are added, modified or deleted by this process.
In target-decoy competition (TDC), both sets of results are pooled and the best match for a spectrum is selected the by highest score. TDC may give different results each time a search is repeated if you use a randomised decoy algorithm, which is why Mascot uses the simpler empirical null approach.
To turn off decoy searching, use one of these options:
- When you submit the search, set target FDR to “(unset)”. This means Mascot will not automatically adjust the score threshold based on the decoy results.
- If you submit the search from Mascot Daemon, just uncheck the Decoy checkbox in the parameter editor.
- If you submit the search from the web browser, go to the Mascot configuration editor and set the option AlwaysEnableAutoDecoySearch to 0 (disabled). You can then uncheck the Decoy checkbox in the search form. To re-enable, set the option to 1.
Target FDR
When you submit a search, you can set the desired target FDR. The setting can also be changed in format controls in the Protein Family Summary report.
The choice of values is a global setting in the options section of mascot.dat.
TargetFDRPerCent 0.1, 0.2, 0.5, 1+, 2, 5
The numbers are percentages and the plus sign doesn’t appear in the drop down list, it indicates the default selection. Clicking on the Decoy link will load a report for the decoy search, just as if it was a separate search of a decoy database.
Decoy statistics are always calculated from all matches. Changing the number of protein hits to be displayed or setting a cut-off on the ions score or expect value will have no effect. Sometimes, it will not be possible to achieve the requested false discovery rate; the significance threshold will be pushed to its limit and a warning displayed.
Peptide FDR
Mascot reports two false discovery rates at peptide level: sequence FDR and PSM FDR. The drop-down menu switches between these.
Sequence FDR counts sequences. Only the highest scoring match to a given sequence is counted, and charge state and modification state are ignored. Note that some software packages report a different kind of peptide FDR, which includes modifications and sometimes charge state. Sequence FDR is slightly more conservative than these.
PSM FDR counts peptide-spectrum matches. All significant peptide sequence matches are counted. For small DDA searches, there is not much difference between PSM FDR and sequence FDR. However, for large searches and especially if you have many duplicate matches, we recommend sequence FDR as it filters out a lot of false positives. Controlling sequence FDR also directly controls protein FDR.
In the example screenshot, target FDR is to 1% sequence FDR, which gives 27848 unique sequences. This is achieved by Mascot by setting the significance threshold to p<0.007771. If you keep the threshold the same but switch to the count of PSMs, there are 84450 significant PSMs at 0.47% PSM FDR.

The PSM FDR is smaller than the sequence FDR, because this data set has many duplicate matches to the same peptides.
Protein FDR
Protein false discovery rate is estimated using a MAYU type of approach. Protein FDR estimation is always convoluted with protein inference, so the behaviour is not obvious.
Only peptide sequence matches (PSMs) with significant scores are used as evidence for proteins. Proteins with shared PSMs are grouped into families. Each distinct family member contains at least one unique peptide sequence, not shared with other family members.
The protein count used for FDR is a count of family members. That is, if the report contains 2 families, one with 4 members and the other with a single member, this counts as a total of 5 proteins. Same-set, sub-set and intersection proteins are not counted.
A protein identification is considered to be true positive if it contains at least one true positive PSM. A protein is a false positive only when all of its PSMs are false positives.
The number of wholly false proteins is estimated by taking the count of decoy proteins and using a hypergeometric distribution to estimate how many of the target proteins contain both true and false PSMs, hence should be counted as true. This correction is important whenever a large fraction of entries may be true hits.
The main differences from the MAYU approach are that we do not make a separate estimate of the FDR for one-hit wonders and we do not partition the database by protein size. We use a simpler estimate for the number of false proteins in the target database, based on the assumption that the number of decoy proteins never reaches a significant proportion of the database size.
Usually, database redundancy causes protein inference ambiguity, meaning we could account for the PSM evidence using several sets of proteins. A protein FDR of 1% only tells us that 1% of the proteins listed are wholly false. This doesn’t mean the other 99% are “correct”. In particular, where there are same-set proteins, we cannot say which one is “correct”.
Relationship between protein FDR and peptide FDR
Protein FDR is controlled by peptide FDR and the minimum count of significant sequences.
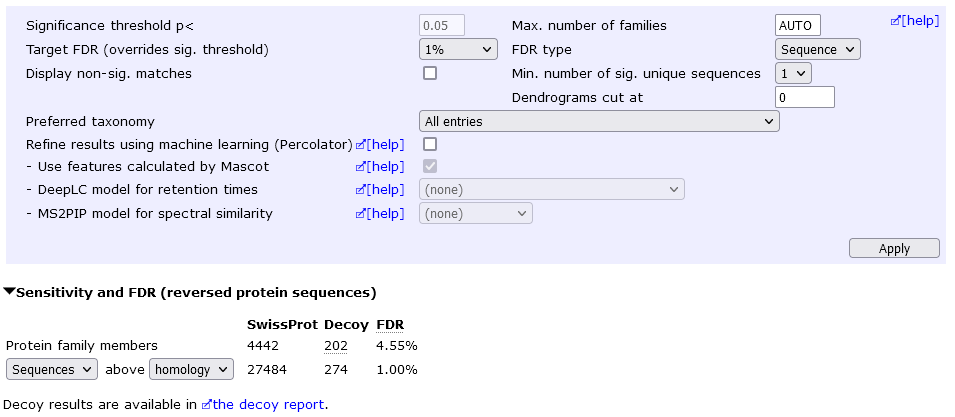
In the below example, the sequence FDR is 1%, which yields 4442 protein hits at 4.55% protein FDR.
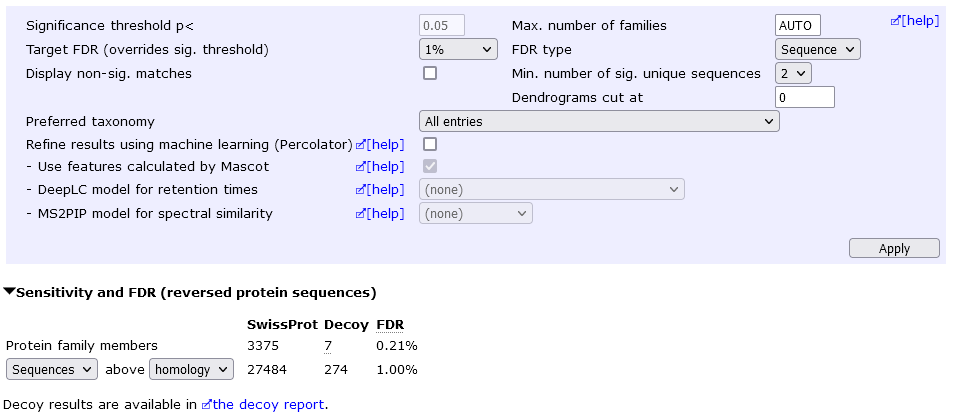
We can eliminate "one-hit wonders" by setting the Min. number of sig. unique sequences to 2 (then choose Apply). This yields 3375 protein hits at 0.2% protein FDR.
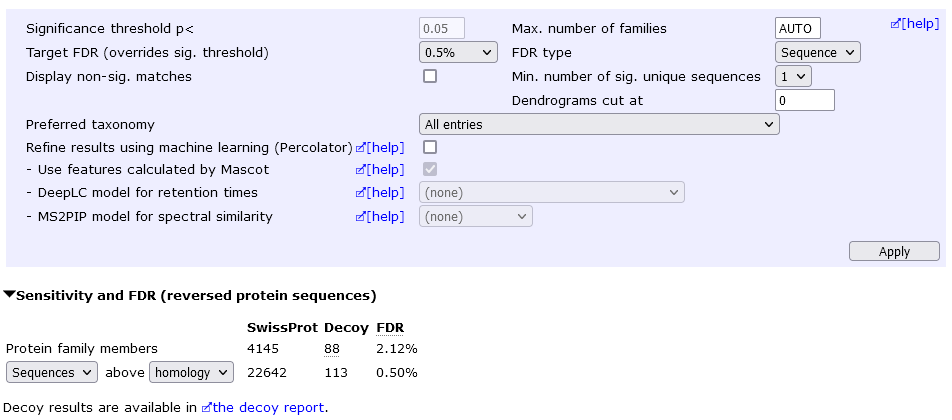
However, you may be able to report a lot more proteins at an acceptable FDR by keeping the "one-hit wonders" and setting a lower peptide FDR. For example, allowing one-hit wonders but setting sequence FDR to 0.5% yields 4145 protein hits at 2.12% protein FDR.
The combination of peptide FDR and Min. number of sig. unique sequences that gives the best sensitivity for a given FDR will be data-set dependant, so use trial and error.
See also the tutorial Creating a list of confidently identified proteins.Peptide Mass Fingerprint FDR
Conventionally, a decoy database search is only used for validating searches of MS/MS data. It is not possible to get a false discovery rate for a peptide mass fingerprint, but it can be informative to see the result of repeating a PMF search against a decoy database, especially if the match from the target database is close to the significance threshold, or if there is reason to think the experimental values or search parameters may be producing a false positive.
This screenshot shows an example of the decoy report for a PMF search:

What makes a good decoy database?
There are actually two target-decoy search strategies: concatenated database and independent searches.
The Gygi group (Elias et al., Nature Methods 2 667-675, 2005) advocate searching a database in which the target and decoy sequences have been concatenated. This means that you will only record a false positive when a match from the decoy sequences is better than any match from the target sequences. This is also known as target-decoy competition (TDC).
A more conservative approach is to search the two databases independently. This models the empirical null distribution more accurately. If the Mascot score threshold for a given spectrum is (say) 40, and we get a match of 60 from the target database and 50 from the decoy database, this would not count as a false positive from a concatenated database, but it would count as a false positive if the two are searched independently.
There is also the question of whether to reverse or randomise. If you simply reverse a sequence, and then do the search without enzyme specificity, you may get a misleading picture of the false positive rate because, sometimes, you will get a mass shift at each end of a reversed peptide that just happens to transform a genuine y series match into a false b series match or vice versa.
Similarly, a reversed database is not suitable for verifying a peptide mass fingerprint score, because half of the tryptic peptide mass values will be unchanged. (Those that have the same residue at the C-terminus and flanking the N-terminus). The main objection to using a randomised database is that the number of distinct peptide sequences in the decoy is likely to be larger than in the target because real protein sequences have a degree of redundancy, which is lost on randomisation.
Algorithms for constructing decoy sequences are described in G. Wang, et al. (2009), "Decoy Methods for Assessing False Positives and False Discovery Rates in Shotgun Proteomics", Anal Chem. 81(1):146-159. The three most widely used are:
- Method 1: reversed protein sequences
- The target protein sequence is reversed, then digested.
- Method 2: reversed peptide sequences
- The target protein sequence is digested, then peptide sequences reversed (keeping C-terminal residue fixed).
- Method 3: randomised protein sequences
- A random protein sequence is generated, where amino acid residue frequency is taken from the database composition. If the target database is nucleic acid, it is the original sequence that is randomised, not the translation. The average amino acid composition of the decoy sequences is the same as the average composition of the target database.
The default for fully specific or semi-specific enzymes, when searching protein sequence databases, is Method 1: reversed protein sequences. This was also the default in Mascot versions 2.4 to 2.8. (Mascot 2.3 defaulted to method 3.)
The default when searching nucleic acid databases is Method 1: reversed protein sequences.
The default for MS/MS searches with enzyme None and all PMF searches is Method 3: randomised protein sequences.
The defaults are specified in the options section of mascot.dat. Refer to the Setup & Installation Manual for further details.
Manual Decoy Search
You can experiment with novel decoy algorithms by turning off automatic target-decoy searching. Mascot Server will then display a checkbox in the search form to enable or disable automatic decoy searching. Create a suitable FASTA file (explained below) and run your database search. You will need to write a suitable script or spreadsheet macro to count the target and decoy matches.
An example Perl script to reverse or randomise database entries can be downloaded here: decoy.pl.gz. Unpack using gzip or 7-Zip.
Note: Windows file associations can cause this file to be unpacked automatically when downloaded using Microsoft Internet Explorer on a Windows PC. If you cannot open the file in 7-Zip, try to open it in a text editor like WordPad. If it looks like text, then it has been unpacked, and you only need to rename the file to decoy.pl.
Execute without arguments to get the following instructions.
Usage: decoy.pl [--random] [--append] [--keep_accessions] input.fasta [output.fasta]
- If –random is specified, the output entries will be random sequences with the same average amino acid composition as the input database. Otherwise, the output entries will be created by reversing the input sequences, (faster, but not suitable for PMF or no-enzyme searches).
- If –append is specified, the new entries will be appended to the input database. Otherwise, a separate decoy database file will be created.
- If –keep_accessions is specified, the original accession strings will be retained. This is necessary if you want to use taxonomy and the taxonomy is created using the accessions, (e.g. NCBI gi2taxid). Otherwise, the string ###REV### or ###RND### is prefixed to each original accession string.
- You cannot specify both –append and –keep_accessions.
- An output path must be supplied unless –append is specified.
- If the database is nucleic acid, no need to specify –random. A simple reversal will effectively randomise the translated proteins
Title line processing assumes that the accession string is between the ">" character and the first white space. If this is not the case, you may need to edit the script to make it usable. If creating a concatenated database, the Mascot parse rules will probably need to be rules 4 and 5 if they are to work for both original and decoy entries. This makes it difficult to configure taxonomy.
The Mascot report scripts cannot display the match counts and FDR after a manual decoy search. One option is to export the results to Excel using the custom CSV format. To avoid outputting duplicate matches when a query matches more than one protein, make sure to set the number of hits to 1, include the unassigned list, and delete any matches with rank greater than 1.
If using a concatenated database, an easier alternative is to use a simple Perl script that can be downloaded here: fdr_stats.pl.gz. Unpack using gzip or 7-Zip.
Copy the script to the Mascot bin directory and execute without arguments to get the following usage instructions:
Output counts of matches for a specified FDR
The program must be run from the mascot bin directory
Usage: fdr_stats.pl fdr_goal thresh_type decoy_string result_file [debug]
Example: fdr_stats.pl 0.01 homology "DECOY_" ../data/20111213/F123456.dat
fdr_goal is the desired peptide FDR (enter 0.01 for 1%)
thresh_type is either identity or homology
decoy_string is the substring in a protein accession that identifies a decoy entry
result_file is the path to a Mascot result file
add optional final argument "debug" to get details of all matches (tab separated)