Identifying peptides from chimeric spectra (DDA, DIA)
Many traditional search engines assume the MS/MS spectrum of a peptide is produced by a single precursor. This is often true when you run a single-species tryptic digest in data dependent acquisition (DDA) mode with a narrow isolation window. Once you change the acquisition strategy or analyse a complex mixture, like an environmental sample, it’s possible for two or more precursors to be selected for fragmentation at the same time, producing a chimeric spectrum. Mascot Server has supported identifying peptides from chimeric spectra since version 2.5, but it doesn’t seem to be widely known among users. Read on for tips how to get started.
Submitting a chimeric spectrum search
When you submit peak lists to Mascot Server, most often this is done as a Mascot Generic Format (MGF) file. A very basic peak list looks like:
BEGIN IONS TITLE=Spectrum 2 PEPMASS=896.05 25674.3 3+ SCANS=123 345.10 237 ... END IONS
The PEPMASS line specifies the precursor m/z (896.05), intensity (25674.3) and charge (3+). If you know that multiple precursors were fragmented into the same spectrum, just add another PEPMASS line:
BEGIN IONS TITLE=Spectrum 2 PEPMASS=896.05 25674.3 3+ PEPMASS=896.43 1084.9 3+ SCANS=123 345.10 237 ... END IONS
If your peak picking software doesn’t produce MGF files with multiple PEPMASS lines per peak list, try Mascot Distiller.
Now, when Mascot reads the peak list, two things happen. First, the peak list is split into two subsidiary queries, one per PEPMASS line, where each query gets a copy of the original peak list. Second, Mascot links the subsidiary queries using a running index, called the source index. With this preparation done, the database search proceeds normally.
During the database search, peptides are matched and scored to subsidiary queries independently. Mascot treats the unknown fragment peaks from the other precursor as unexplained noise. In particular, Mascot does not try to remove or attenuate interfering fragment peaks, which some software tools do in an effort to boost the match score. We feel it’s not only difficult to justify from a combinatorial point of view, but it can also be actively misleading if the precursors share any fragment peaks.
At the end of the search, Mascot records the source index in the query sections of the results file, which can be used by downstream tools to detect chimeric spectra. No other processing specific to chimeric spectra is currently done by the search engine. If your peak picking software inserts the scan number in the MGF file as the SCANS line (Mascot Distiller does), these can be used for grouping chimeric matches. Otherwise you can use the source index.
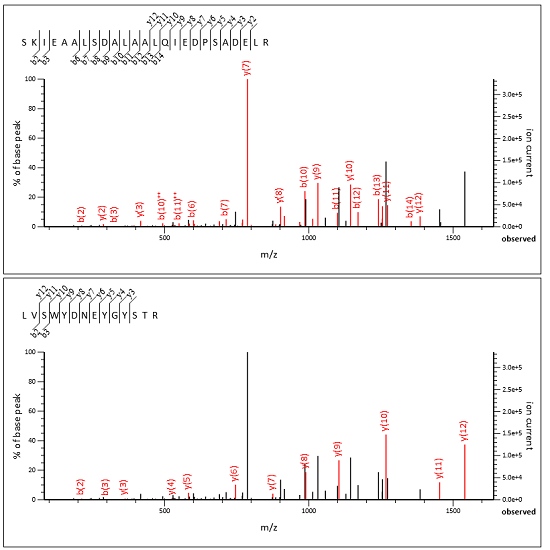
We’ve added a new help page, Chimeric spectra, which contains background and the full details. Below is a screenshot of an MS/MS spectrum split into two subsidiary queries, taken from an example yeast DDA data set, where the two peptide matches jointly explain all the important peaks.
Advantages and limitations
The Mascot approach has advantages despite (or because of!) its simplicity. Most importantly, a chimeric spectrum match must have unambiguous MS/MS evidence in order to get a good Mascot score. We believe you must be able to inspect the spectrum and convince yourself the peptide is truly there, regardless of any external factors like retention time or ion mobility matching. How else would you be able to trust that the software is working correctly? And, why else would you acquire MS/MS spectra in the first place?
On the flip side, although Mascot scoring is tolerant to noise, it becomes more difficult to get a good match as you increase the number of precursors. Mascot can usually identify the primary (most intense) precursor and a secondary, weaker peptide. Sometimes it can identify a third or fourth peptide. As you add fragments from more and more precursors, the score for the weaker peptides approaches zero.
Tip: chimericity report
The reporting in the current version of Mascot Server does not provide a good way to find and navigate chimeric matches. We’ve created a basic chimericity report script that you can download and use with any version of Mascot. The script summarises the number of precursors per spectrum in the peak lists, as well as provides a breakdown of the number of significant peptide matches to chimeric spectra of increasing chimericity.
Tip: change decoy type from reverse to random
We always recommend running a target-decoy search to estimate the false discovery rate in shotgun proteomics experiments and allow Percolator rescoring. However, it isn’t true that “you just need some decoys” for Percolator to work. The decoy matches have to be an accurate model of the false (or “null”) matches in the target database. If decoy sequences have a lower success rate against chimeric spectra than false target sequences, it causes the FDR to be underestimated. If you then feed these poor quality decoys to Percolator, it may exacerbate the problem.
Mascot’s automatic decoy search defaults to reversed protein sequences. After inspecting many chimeric data sets, we have noticed that reversed decoys struggle to get a good match against chimeric spectra as spectral complexity increases. We recommend changing DecoyTypeSpecific from 1 (reverse) to 3 (random) in Mascot options when you run chimeric spectrum searches.
We ran the example yeast DDA data set using reversed and random decoys, summarised in the table below.
| Decoy type | Sig. threshold | Target PSMs | Decoy PSMs | FDR |
|---|---|---|---|---|
| reverse | 0.05 | 45506 | 179 | 0.39% |
| random | 0.05 | 45506 | 341 | 0.75% |
First one looks better, right? Actually, the low FDR is the result of decoy matches scoring poorly against chimeric spectra, which this data set has in plenty. Repeating the search with random decoy sequences doesn’t change the number of target matches, but it does make a better FDR estimate. We’re not fully satisfied with randomised decoys either and will try to improve the behaviour in a future release.
Tip: data independent acquisition (DIA)
In data independent acquisition (DIA), the isolation window is much wider and there is no intensity-based precursor selection. All precursors within a mass range are fragmented simultaneously, producing highly chimeric MS/MS spectra.
DIA strategies can be broadly divided into narrow window (up to 8 m/z) and wide window (> 8 m/z) based on the selected isolation window. Mascot Server is not currently able to process wide window DIA data in a useful way, as these can have dozens of precursors per spectrum.
However, narrow window DIA spectra tend to have only a few precursors per spectrum, and their complexity is similar enough to chimeric DDA spectra that Mascot can be used successfully. We will provide a concrete example of processing a narrow window DIA data set with Mascot in a later blog article.
Keywords: chimeric spectra, FDR