General approach to modifications
Most protein samples exhibit some degree of modification.
There are the "natural" post-translational modifications, such as phosphorylation and glycosylation. There are the accidental modifications which are artefacts of sample handling, such as oxidation of methionine. Finally, there are the modifications deliberately introduced during sample work-up, such as cysteine derivatisation. In most cases, it is only the deliberate modifications which are known about for certain at the time of doing a search.
It might be assumed that the search software could allow for those modifications which are described in sequence entry annotations. However, in public sequence databases, many post-translational modifications are not specified in a way which can be readily translated into specific mass differences. For example, noting that a residue is an actual or potential glycosylation site is not much help. Even a simple modification, such as phosphorylation, is rarely quantitative, so that it would be necessary to include mass values for all permutations of occupied and unoccupied sites.
And, of course, protein sequences derived translated from nucleotide sequences contain no information on post-translational modifications.
Mascot solves the problem by allowing modifications to be specified in two different ways: fixed modifications and variable modifications. Quantitation methods support an additional mode, exclusive modifications.
Fixed modifications (quantitative)
Fixed modifications are those which are always present, sometimes called quantitative modifications.
Fixed modifications are applied universally, to every instance of the specified residue or terminus. There is no computational overhead associated with a fixed modification; it is simply equivalent to using a different mass for the modified residue or terminus. For example, selecting Carboxymethyl (C) as a fixed modification means that all calculations will use 161 Da as the mass of cysteine.
In general, any modification can be specified as a fixed mod. There are two restrictions: A fixed modification cannot have more than one fragment neutral loss. You also cannot select a fixed modification with protein N-terminal or protein C-terminal specificity (every peptide has an N-terminus and a C-terminus, but most peptides are not protein terminal).
Variable modifications (qualitative)
Variable modifications are those which may or may not be present, sometimes called qualitative modifications. Common examples are oxidation of methionine and phosphorylation.
Mascot tests all possible arrangements of variable modifications to find the best match. For example, if Oxidation (M) is selected as a variable modification, and a peptide contains 3 methionines, Mascot will test for a match with the experimental data for that peptide sequence containing 0, 1, 2, or 3 oxidised methionine residues. This greatly increases the complexity of a search, resulting in longer search times and reduced specificity, so variable modifications should be used sparingly.
Exclusive modifications
Exclusive modifications can be thought of as multiple-choice fixed modifications. In many quantitation experiments, separate samples are derivatised then pooled. Thus, a given peptide may carry one or the other set of modifications, but never a mixture of both. Some people use the term "binary" for this type of specificity. We prefer exclusive because binary implies only two possibilities. The value of exclusive modifications is that they keep the search space small, which avoids the combinatorial explosion that can occur with too many variable modifications.
Exclusive modifications can only be specified as part of a quantitation method; they cannot be selected in the search form. There is a related parameter called Constrain search. If this is set true, exclusive modifications are treated as a choice of fixed modifications during the search. This makes the search space small so that the search is fast and the score thresholds are kept low. If Constrain search is false, exclusive modifications are treated as variable during the search, and it is only during quantitation that they are treated as a choice of fixed modifications. This can be useful if you suspect chemistry problems, and want to see matches to peptides that are only partially modified.
Sometimes, you may be forced to set Constrain search to false because Mascot doesn’t allow one or more of the modifications to be fixed. An example would be a protein N-terminus modification or a modification with multiple neutral losses. Only residue and peptide terminus mods with zero or one neutral losses can be fixed.
Identifying unsuspected modifications
Variable modifications to search for must be specified up front, as search parameters. When there is uncertainty about the degree or identity of modifications, use the error tolerant search to identify unsuspected modifications.
Limits on the number of variable modifications allowed in a search
There are separate limits on the number of variable modifications allowed in a search.
MaxVarMods is a soft limit that defaults to 9. This specifies the maximum number of variable modifications selected in the search form. The maximum for MaxVarMods is 32. You may have to increase this limit when searching for intact crosslinks combined with monolinks and variable modifications, but otherwise the limit of 9 is reasonable.
MaxEtVarMods is a soft limit that defaults to 2. This specifies the maximum number of variable modifications selected in the search form in an error tolerant search. The limit of 2 can be a bit restrictive, so increasing it to 4 or 6 is justified.
If Mascot Security is disabled, these are global limits set in the options section of mascot.dat. If Mascot Security is enabled, these limits can also be set at security group level, in which case changing the mascot.dat settings may have no effect.
The combinatorial explosion resulting from too many variable modifications is a real problem, leading to searches that take forever and a serious loss of sensitivity. Typically, you pick up a few additional matches to highly modified peptides but lose a much larger number of matches to peptides from low level proteins because of the increase in the score required for a match to be significant. On the other hand, a simple limit on the total number of variable modifications is a crude tool, because different modifications can have very different effects. Modifications that only apply to a protein terminus, such as Acetyl (Protein N-term), or to a specific residue at a peptide terminus, such as Gln->pyro-Glu (N-term Q), cause a negligible increase in the search space. The dramatic effects come from modifications that apply to multiple residues, independent of location, such as Phospho (ST) or Methyl (DE). These are the ones that need to be used very sparingly.
Limits on the number of variable modifications allowed in a peptide
When a search contains many variable modifications, there may be a large number of possible modification states for a candidate peptide that agree with the experimental mass. It is necessary to place an upper limit on the number of possibilities that are tested and scored. Otherwise searches would become unacceptably slow. To illustrate how these limits work, consider an example:
Variable modifications selected for the search: Acetyl (K), Acetyl (N-term), Methyl (K), Dimethyl (K), Trimethyl (K)
Candidate peptide: QLATKAARKSAPSTGGVKKPHRYKPGTVALK with m/z corresponding to a modification delta of 126 Da
Assume that the mass accuracy is +/- 0.2 Da, so that we cannot tell whether a peptide is modified by Acetyl (42.01 Da) or Trimethyl (42.05 Da)
Limits set in mascot.dat:
- A limit on the number of distinct varmods found on a single peptide: MaxPepNumVarMods=3
(3 x Acetyl (K) is a single distinct varmod, 1 x Acetyl (N-term) + 2 x Acetyl (K) is two distinct varmods)- A limit on the number of modified sites found on a single peptide: MaxPepNumModifiedSites=5
- A limit on the number of arrangements of an individual varmod composition: MaxPepModArrangements=64
The first step is to enumerate all possible varmod compositions that fit to the experimental precursor mass of the candidate peptide. The constraints on this list are MaxPepNumVarMods, MaxPepNumModifiedSites, and the fact that the candidate peptide contains six K residues and one N-term. For example, the total delta of 126 could be any of the following compositions:
- 3 x Acetyl (K)
- 3 x Trimethyl (K)
- 1 x Acetyl (N-term), 2 x Acetyl (K)
- 1 x Acetyl (N-term), 1 x Acetyl (K), 1 x Trimethyl (K)
- 1 x Acetyl (N-term), 2 x Trimethyl (K)
- 2 x Acetyl (K), 3 x Methyl (K)
- and many others
But, it could not be one of these compositions, even though they add up to the correct delta mass:
- 2 x Acetyl (N-term), 1 x Acetyl (K) – not allowed, only one N-term available
- 3 x Methyl (K), 3 x Dimethyl (K) – not allowed, exceeds MaxPepNumModifiedSites
- 1 x Acetyl (K), 1 x Methyl (K), 1 x Dimethyl (K), 1 x Trimethyl (K) – not allowed, exceeds MaxPepNumVarMods
For each composition that fits the required delta mass, multiple arrangements may be possible. Different arrangements have the same peptide mass but will give rise to differences in the MS/MS spectrum. For example, there are 20 possible arrangements of 3 x Acetyl on 6 x K, here shown schematically:
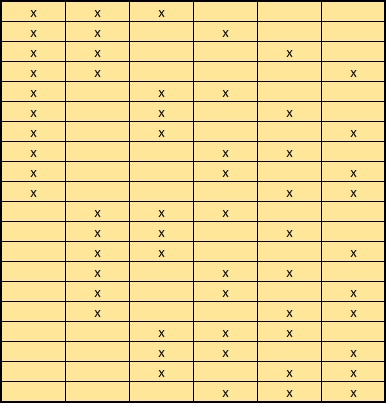
If the number of arrangements of an individual composition is less than MaxPepModArrangements, all can be tested and the highest scoring match reported. If the number of arrangements is greater than MaxPepModArrangements, arrangements are tested in random order, so that the entire space of possible arrangements is sampled before reaching the limit.
For example, there are 180 possible arrangements of 1 x Acetyl (K) + 2 x Methyl (K) + 2 x Dimethyl (K), of which only 64 would be tested. Even if this happened to be the correct composition, there is only a 1 in 3 chance that the perfectly correct arrangement will be one of those that are scored. Even if the MS/MS spectrum is of very high quality, the reported match is likely to be a nearly correct arrangement rather than the perfectly correct arrangement. If finding the best possible match is important, and this was a possible composition, you would need to increase MaxPepModArrangements and accept that the search would be slower.
Whatever the limits, for best speed and specificity, it is essential to minimise the number of variable modifications included in a search. For example, if the mass accuracy does not allow Acetyl (K) and Trimethyl (K) to be distinguished, one should be dropped. If the interest is in post-translational modifications, select Acetyl (Protein N-term), not Acetyl (N-term).
Multiple modifications at a single site
Outside of quantitation, there aren’t many occasions when you need to match peptides with multiple modifications at a single residue or terminus. In most cases, multiple modifications don’t occur because of chemistry, e.g. you can’t have Carbamidomethyl (C) and Propionamide (C) on the same cysteine. In other cases, incremental modifications are handled via separate modifications, e.g. Methyl (K), Dimethyl (K), and Trimethyl (K).
The picture for SILAC quantitation is different because SILAC is implemented using modifications. One option is to manually combine the modification definitions to create a new modification. For example, in Unimod, you’ll find modifications such as Label:13C(4)+Oxidation and Label:13C(6)+Acetyl. This is unsatisfactory because it doubles the number of variable modifications required.
Mascot supports multiple modifications at the same site, but only in the context of a quantitation method. The restrictions are:
- Two modifications at a single site are allowed, but only one of these can be a variable modification. The other must be an exclusive modification, defined in a quantitation method, such as a SILAC label.
- Constrain search must be set true in the quantitation method.
- The search must include the parameter MULTI_SITE_MODS=1.
For example, in a 3 component SILAC experiment, using R+6 and R+10, if we had a match to this peptide:
K.QMEQISQFLQAAER.Y + Oxidation (M); Label:13C(6) (R)
It would partner with these, whether or not we also got matches for them:
K.QMEQISQFLQAAER.Y + Oxidation (M)
K.QMEQISQFLQAAER.Y + Oxidation (M); Label:13C(6)15N(4) (R)
It would not partner with:
K.QMEQISQFLQAAER.Y + Oxidation (M); Phospho (ST)
K.QMEQISQFLQAAER.Y + Label:13C(6)15N(4) (R)
etc.
Be a little bit careful when performing non-SILAC quantitation. For example, if you are using dimethyl labelling, and you specify other variable modifications for K or N-term, there is no chemical intelligence in Mascot to decide whether the variable modification and the dimethyl label could both apply to the same site. If the peptide mass fits to the combination, it will try both together. You shouldn’t get a significant match, of course, if the peptide doesn’t exist, but if you start digging in the low scoring junk, you might find some unlikely looking combinations.
Neutral Losses
Mascot supports four types of neutral loss.
Scoring: A neutral loss from the MS/MS fragments. The resultant ions are considered for scoring, e.g. y-98 or b-98 for phosphopeptides. There can be up to 10 scoring neutral losses.
Satellite: A neutral loss specified as satellite is never considered for scoring. None of the standard modifications in Unimod currently have satellite neutral losses.
Peptide: A neutral loss from the intact peptide precursor.
Required Peptide: A required peptide neutral loss must be present in the spectrum.
During a database search, Mascot treats neutral losses in the following way:
- Required peptide NL is a filter. If there is no peak for the neutral loss, the peptide is not matched at all. This carries some risk, because a perfectly good match could be rejected if this peak was missing.
- Peptide neutral loss peaks are matched but not explicitly scored. A matching peak is removed from the list of noise peaks, which improves the score.
- If there are multiple fragment neutral losses, Mascot iterates through the Scoring ones. The loss that gives the highest score is chosen, and all the other neutral losses are treated as Satellite.
- Finally, Satellite neutral loss peaks are matched but not explicitly scored. A matching peak is removed from the list of noise peaks, which improves the score.
Example: phosphorylation
Phosphorylation is one of the most interesting and studied modifications. It is also one of the most challenging for database searching, because of these factors:
- Site heterogeneity
- 3 fragmentation channels
- intact fragments
- neutral loss of HPO3 (80 Da)
- neutral loss of H3PO4 (98 Da)
- Can occur at STY – ~16% of residues.
In the default phosphorylation modifications derived from Unimod, pY fragments always stay intact, while pS and pT fragments can stay intact or can lose 98.
This is not a hard and fast rule, and sometimes a loss of 80 is also observed. However, this is not included in the definition because it is identical to the delta of the original modification. Allowing for the possibility of 80 Da neutral loss introduces ambiguity as to the site of the modification when there are multiple potential phosphorylation sites in a peptide. For example, this match to pTESPATAAETASEELDNR gets a score of 115:
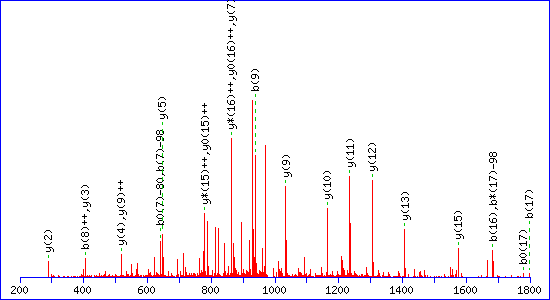
If a neutral loss of 80 Da is allowed, the score for a match to TESPATAAETApSEELDNR is almost as high, 92:
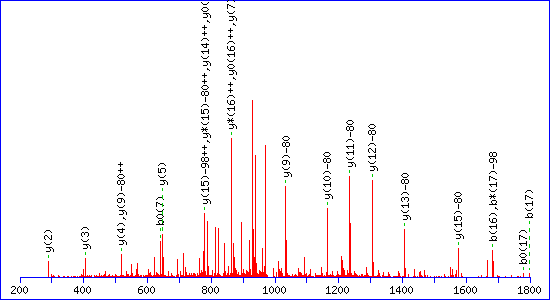
The reason is clear. The matching peaks are all y ions, so the point of modification can be shifted towards the C-terminus by swapping the matching series from y to y-80. Without the availability of an 80 Da loss, the score for the second match drops to 29.
It has often been observed that the neutral loss from the precursor can be an excellent guide to the identity of the phosphorylated residue. If a strong loss of 98 Da is observed, then the expectation is pS or pT. If no neutral loss, then pY. In Mascot, one or more precursor neutral losses can be specified. They can also be made "required", which means that the peak must be present in the spectrum. This carries some risk, because a perfectly good match could be rejected if this peak happened to be missing.
Configuration
Unimod
The list of modifications used by Mascot is taken directly from the Unimod database. For further details of individual modifications, please refer to Unimod. Note that Unimod is a community supported resource. If you want to add a new modification to Unimod, you can do so, and you then become the curator of the new record. The Mascot modifications list on the public web site is updated from Unimod regularly.
By default, only selected modifications are displayed in the Mascot search form. If you want to see the complete list, you must go to the search form defaults page and tick the checkbox for ‘Show all mods.’.
In Unimod, both amino acid residues and modifications are defined in terms of their elemental composition. This is important for metabolic labelling, in which the isotopic label is present throughout the peptide backbone.
Adding your own modifications
For an in-house Mascot Server, if you don’t see the modification you require, first check the public Unimod database. If the modification is there, you just need to update the Unimod database on your local Mascot Server (go to the Configuration Editor, Modifications and click ‘Check Unimod’). If the modification is not in Unimod, you can either add it to the public Unimod database and wait for the downloadable file to be rebuilt or use the browser-based Configuration Editor to add it locally:
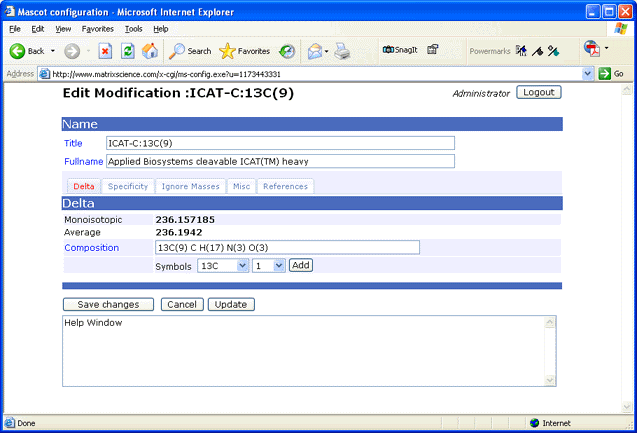
You can copy and make local edits to Unimod definitions. You can also define modifications specific to your experiment.
Other useful databases of modifications for reference purposes are RESID, a database contains detailed descriptions of many post-translational modifications, and PSI-MOD.
Grouping of modifications
Some modifications have multiple specificities, e.g. Phospho (ST) and Methyl (DE). This is achieved by giving the specificities the same non-zero group number in the modification definition. For example, Phospho S and T specificities are both group 1, while Y is group 2. The rules governing grouping are:
- Only simple residue specificities can be grouped (Position = Anywhere). You cannot group specificities such N-term Q or group residue with terminus specificities.
- The neutral loss definitions must be identical. This is why Phospho (Y) cannot be grouped with Phospho (ST).
Fixed and variable modifications with the same specificity
You cannot have two fixed modifications in a search with the same specificity. That is, you cannot have both Carbamidomethyl (C) and Propionamide (C) as fixed modifications.
If a search contains a variable modification with the same specificity as a fixed modification, the variable can displace the fixed mod. For example, if Carbamidomethyl (C) and Propionamide (C) were both variable modifications, there would be three possibilities for each cysteine: modified with Carbamidomethyl, modified with Propionamide, or unmodified. If Carbamidomethyl (C) was fixed and Propionamide (C) was variable, then you would only see one or the other; you would never see an unmodified cysteine. If you also want to match peptides with unmodified C, both modifications need to be variable.
A frequently reported problem is getting the error message "Modification conflict" when submitting an iTRAQ or TMT search. This is because the labels are specified in the quantitation method as fixed modifications, and you have either selected them a second time in the search form or chosen another fixed modification with either K or N-term specificity.

