Quantitation overview
Many different approaches to protein quantitation using mass spectrometry data have been described in the literature. For a short, recent review, see Ong, S. E. and Mann, M., Mass spectrometry-based proteomics turns quantitative, Nature Chemical Biology 1 252-262 (2005). In terms of the "mechanics" of their implementation, most of the popular approaches can be classified into a relatively small number of protocols:
- Reporter: Quantitation based on the relative intensities of fragment peaks at fixed m/z values within an MS/MS spectrum. For example, iTRAQ and Tandem Mass Tags
- Precursor: Quantitation based on the relative intensities of extracted ion chromatograms (XICs) for precursors within a single data set. The approach can be used with any chemistry that creates a precursor mass shift. For example, 18O, AQUA, ICAT, ICPL, Metabolic, SILAC, etc., etc.
- Multiplex: Quantitation based on the relative intensities of sequence ion fragment peaks within an MS/MS spectrum. This is a novel approach, which can be used with labels located at the peptide terminus, such as 18O or SILAC at K or R in combination with tryptic digestion.
- Replicate: Label free quantitation (LFQ) based on the relative intensities of extracted ion chromatograms (XICs) for precursors in multiple data sets aligned using mass and elution time.
- emPAI: Label free quantitation (LFQ) for the proteins in a mixture based on protein coverage by the peptide matches in a database search result.
- Average: Label free quantitation (LFQ) for the proteins in a mixture based on the application of a rule to the intensities of extracted ion chromatograms (XICs) for the peptide matches in a database search result.
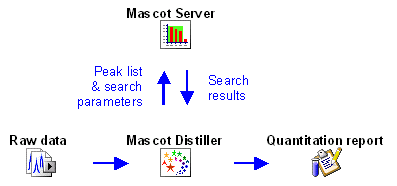
Some protocols can be fully implemented within a Mascot result report because all the necessary information is present in the peak list. These protocols are Reporter, Multiplex, and emPAI. In fact, emPAI is "always on", and will be reported whenever an MS/MS search contains at least 100 spectra.
The other three protocols require additional information from the raw data file, either because it is necessary to integrate the elution profile of each precursor peptide or because information is required for precursor peptides that were not used to trigger MS/MS scans, so are missing from the peak list. So, for Precursor, Replicate, and Average, the quantitation report is generated in Mascot Distiller, which has access to both the Mascot search results and the raw data.
Besides the choice of protocol, there are a large number of other choices and parameters associated with searching, processing, and reporting of quantitation data. These choices and parameters are necessary to provide sufficient flexibility and control, yet it would be undesirable to expose all of them in the search form. The solution is to encapsulate all the settings for a quantitation experiment into a named quantitation method. This means that quantitation support requires just a single control in the search form:

The set of quantitation methods is defined by an XML configuration file, called quantitation.xml. As with other configuration files, this file lives on the Mascot Server and is downloaded by Mascot Distiller and other clients as required. Brave souls may choose to edit the XML file directly, but a more friendly interface is provided by a browser based configuration editor.
Modifications
Mascot takes its modification definitions direct from an XML representation of the Unimod database. To update the local definitions, simply download the latest XML file from the Unimod help page.
In Unimod, both amino acid residues and modifications are defined in terms of their elemental composition. This is important for metabolic labelling, in which the isotopic label is present throughout the peptide backbone.
Unimod also provides a framework for including local definitions of modifications within a quantitation method. For example, the multiplex method may require that a modification has two neutral losses. One of 0 Da and one corresponding to the complete modification moiety, so that a mixed spectrum, containing both labelled and unlabelled peptides, can be matched with a good score. It would be confusing to have such an artificial modification appearing in Unimod, so the preference is to define it within the quantitation method.
In the context of a quantitation method, Mascot now supports exclusive modifications. A group of exclusive modifications can be thought of as a choice of fixed modifications. In many quantitation experiments, separate samples are derivatised then pooled. Thus, a given peptide may carry one or the other set of modifications, but never a mixture of both. Some people use the term "binary" for this type of specificity. We prefer exclusive because binary implies only two possibilities. A SILAC experiment might have three or four different labels which will never be mixed on a single peptide. One of reasons for introducing this new type of modification is that variable modifications greatly increase the size of the search space, because all of the possible permutations and combinations of modified and unmodified residues have to be explored. As the search space becomes larger, the search takes longer and the score threshold increases, making it more difficult to get significant matches from marginal spectra. By treating labels as a choice of fixed modifications, we avoid this combinatorial explosion.
Key concepts
Two key concepts have already been introduced. A quantitation method encapsulates all the settings for searching, processing, and reporting of quantitation data. The "mechanics" of the method are specified by the protocol. These keywords represent concepts, and also structures in the configuration file. You will see these words used consistently, (we hope), in this help, in the browser based configuration editor, and in the XML configuration file.
Another important concept and keyword is a component. This is the characteristic property of a peptide that identifies its origin in the sample mixture. For example, in a SILAC experiment, one component might be unmodified peptides while another component is peptides modified with Label:13C(6) on arginine or lysine. If the protocol was reporter, then a component would be identified by a reporter ion m/z value. If it was a metabolic labelling experiment, then one component might be 14N peptides and the other 15N peptides. In a label free experiment, using the replicate protocol, each data file would be a component.
If the one component was called light and the second heavy, we might want a report to list the ratio of heavy over light, or maybe light over heavy. This is specified as a report ratio and is not limited to two components. In an 18O experiment, for example, you might want to report the ratio of (18O1 + 18O2) / 18O0. Both the numerator and denominator of a reported ratio can be linear combinations of components. For example, (0.5 * A + 0.5 * B – C) / (D + E) … not that we can think of a practical use for something so complicated.
In a quantitation method, modifications are organised into groups, classified as fixed, variable, or exclusive. Modification groups can be defined as variable or exclusive at the component level, where they usually characterise the component. They can also be defined at the method level, but only as fixed or variable. Defining modifications at the method level is a convenience, for modifications that are important to the method, and saves having to choose them in the search form.
The other method level tabs in the configuration editor, which correspond to child elements of the method element in the XML file, are:
- integration: Choices and parameters to control the way in which extracted ion chromatograms are integrated in Mascot Distiller. Also, how to align precursors in the replicate protocol.
- quality: Miscellaneous quality criteria that peptide matches must meet before they can be used for quantitation. The most important is the strength of the peptide match, defined in terms of either a minimum score, a maximum expect value, or the score being at or above either the identity threshold or the homology threshold.
- outliers: When ratios for individual peptide matches are combined into ratios for protein hits, a variety of procedures are available for detecting and rejecting outliers
- normalisation: Whatever the quantitation method, it can be difficult to ensure that each component is treated identically. If it is reasonable to expect that only a minority of the proteins in the sample will be up or down regulated, global normalisation can be applied so as to make the average or median ratios across the entire data set unity. Alternatively, known amounts of exogenous protein or peptide can be spiked into the samples and specified as the basis for normalisation.

