Identifying unsuspected modifications with Error Tolerant search
The Mascot Error Tolerant Search is a two-pass search intended for identifying unsuspected variable modifications, enzyme non-specificity and sequence variants not present in the sequence database.
When to use error tolerant searching
When the results of an MS/MS Ions Search of an LC-MS/MS dataset are reviewed, there will often be a number of spectra that remain unmatched. Assuming that a given MS/MS spectrum contains adequate information, i.e. a reasonable number of fragment ion peaks at usable signal to noise, possible reasons for this failure include:
- Enzyme non-specificity
- Unsuspected chemical & post-translational modifications
- Peptide sequence not in the database
- Underestimated mass measurement error
- Incorrect determination of precursor charge
The Mascot Error Tolerant search addresses the first three issues.
If mass measurement error has been underestimated, this should be apparent from the graphs showing the differences between the calculated and measured mass values in the Peptide View and Protein View reports.
Incorrect determination of precursor charge has to be dealt with during peak detection. If it is not possible to determine the precursor charge reliably, then one option is to generate peak lists for all probable charge states.
Error tolerant searching is only applicable to MS/MS data; it is not possible to perform an error tolerant peptide mass fingerprint. For a truly unknown modification, or a sequence variation of more than a single base or residue, the error tolerant sequence tag is worth investigating.
How to submit an error tolerant search
An automatic error tolerant search is performed by choosing “Automatic second pass search of selected modification classes” in the search form.
The following constraints apply to the other search parameters:
- Enzyme must be fully specific
- There is a reduced soft limit on the number of variable modifications (the limit can be increased)
- Error tolerant searching cannot be combined with quantitation
- Error tolerant searching cannot be combined with crosslinking
- Error tolerant searching cannot currently be combined with refining results with machine learning
- The search cannot include error tolerant sequence tags
The target database can be a protein sequence database or nucleic acid sequences.
We recommend starting with a small database, such as a UniProt proteome or SwissProt. An error tolerant search of a very large database like UniRef100 or NCBIprot is possible, but the search will take extremely long and you will not get many significant matches due to high score thresholds.What Mascot does
A standard, first pass search is performed using the search parameters specified in the form. From the results of the first pass search, all of the database entries that contain one or significant peptide matches are selected for an error tolerant, second pass search.
The automatic error tolerant search is always run as an automatic target-decoy search. If target FDR is (no target), a simple expect value test is performed to assess statistical significance: peptide match expect value < 0.05. If a value is specified for target FDR, the significance threshold for the first pass search is automatically adjusted to achieve this target. In both cases, the significance test determines which queries go forward to the second pass (those without significant matches) and which proteins will be searched (those with one or more significant matches).
At the completion of the second pass search, a single report is generated, combining the results from both passes.
During the error tolerant, second pass search:
- The selected enzyme becomes semi-specific (that is, only one end of a peptide needs to match the cleavage specificity), and the value of the missed cleavage parameter is increased by 1.
- The complete list of modifications is tested, serially.
- For a protein, the set of all possible amino acid substitutions is tested. For a nucleic acid sequence, all single base insertions, deletions, and substitutions are tested.
- Only one of the above is allowed per peptide. That is, an individual peptide can be semi-specific OR have one unsuspected modification OR have one primary sequence mutation.
- If the mass delta of the modification is less than the smaller of the precursor mass tolerance and the fragment mass tolerance, the modification is rejected. This eliminates modifications that are meaningless given the estimated mass error, like Q->K, in most cases.
In the second pass, Mascot skips combinations of modifications that were already searched in the second pass. Mascot also skips fully specific peptide sequences and only tests semi-specific peptides. This means the first and second pass search spaces are disjoint.
Statistical validation
The decoy database for the second pass is constructed in a special way. The target and decoy proteins are treated as pairs. After the first pass search, when proteins are selected, each significant peptide match (whether target or decoy) causes the relevant pair of target and decoy proteins to be selected for the second pass. This means the second-pass decoy database is a combination of decoy protein hits with a significant match, and decoy proteins that did not get a significant match, but whose paired target protein did. Conversely, the second-pass target database is a combination of target protein hits with a significant match, and target proteins whose paired decoy protein got a significant match. The second-pass decoy database has identical size to the second pass target database, and the proteins cover all significant peptide matches (PSMs) from the first pass.
If a query gets a significant match in the first pass search, it is omitted from the second pass. Thus, a query searched in both passes had no matches or only non-significant matches in the first pass. The identity threshold for this query is based on the number of trials from first pass + second pass to reflect the combination of the search spaces. In the second pass, the statistical significance of the error tolerant match is subject to this much stricter threshold.
Because the first and second pass search spaces are disjoint, this means the first and second pass results can be treated independently. When you select a target FDR, thresholding is applied in two steps. First, results from the first pass are thresholded to yield the target FDR. Second, results from the error tolerant pass are thresholded using an independent significance level. Then the two (disjoint) sets of results are merged into a single report. As long as both sets of results have the same FDR, for example 1% FDR, the union of the disjoint sets also has the same FDR.
Selecting classes of modifications
The list of modifications used by Mascot is taken directly from the Unimod database.
The default is to test all the entries on the modifications list serially. For each modification in Unimod, Mascot tries the modification on its own as well as combining it with variable modifications specified as search parameters. For each combination, every permutation is tested within configured limits. For example, if a modification affects serine, and a peptide contains three serines, but has a molecular mass consistent with just two modifications, there are 3 permutations to be tested (110,101,011).
Up to 1 error tolerant modification is allowed per peptide, but there can be several instances of the modification. For example, suppose the error tolerant modification is Methyl (DE) and the peptide is xxDxMxxDExxxxEx, and Oxidation (M) is selected as a variable mod. In the second pass of the search, Mascot tries 1, 2, 3 and 4 instances of Methyl (DE), combined with 0 or 1 instances of Oxidation (M). The combinations that fit the precursor tolerance are matched and scored.
Specifying more than a handful of variable modifications leads to a drastic loss of discrimination, because the number of permutations and combinations increases geometrically with the total fractional abundance of modifiable residues.
You can optionally limit the modifications tested in the second pass by choosing specific modification classes. The modification classes are defined in Unimod and include, for example, chemical derivatives, post-translational modifications and N or O-linked glycosylation.
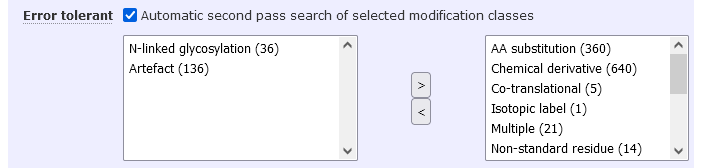
In the above screenshot, only modifications whose class is Artefact or N-linked glycosylation will be searched in the second pass. The search form displays the number of modifications in each class. In this case, a total of 172 error tolerant modifications will be tested in combination with any other variable modifications selected.
More about Sequence Variants
Variations in the primary sequence generally result from variations in the DNA sequence. These may be DNA sequencing errors, they may be mutations or polymorphisms, or they may be more extensive evolutionary changes, because the database entry is not the authentic protein, but a related sequence from a different species.
When searching a nucleic acid database, single base deletions and insertions can be tested in addition to substitutions. The consequences of deletions and insertions cannot be tested for a protein database because they cause a frame shift, which completely changes the amino acid sequence from that point onwards.
Amino acid substitutions in protein sequences are handled like modifications, and the composition and mass changes are taken from Unimod entries.
Tutorial
Please see the tutorial reviewing error tolerant results for a concrete example.
