Mass-tolerant vs Error tolerant
"A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides" in Nature Biotechnology is from Steven Gygi’s lab at Harvard Medical School. It describes the use of a very wide precursor mass tolerance, +/- 500 Da, to identify modified peptides in a Sequest search.
How does this approach, which the authors call an open search, compare with a "conventional" multi-pass search, such as the Mascot error tolerant search? To find out, we downloaded Gygi’s HEK 293 cell data set, consisting of 24 Q-Exactive Orbitrap raw files, from Pride project PXD001468. Mascot Daemon was used to automate peak picking of the files using Mascot Distiller, merge them, and submit an error tolerant search to Mascot Server 2.5.1. The Distiller processing options can be downloaded here. The sequence database was identical to that used in the paper (GRCh37.61.pep.all) and the other search parameters were:
Enzyme : Trypsin/P Fixed modifications : Carbamidomethyl (C) Variable modifications : Oxidation (M) Mass values : Monoisotopic Protein mass : Unrestricted Peptide mass tolerance : ± 5 ppm (# 13C = 1) Fragment mass tolerance : ± 15 ppm Max missed cleavages : 2 Instrument type : ESI-TRAP
A separate search was used to determine the significance threshold to give a peptide FDR of 1% for the first pass search. The most abundant modifications, with more than 1000 instances each, from the open search (as listed in Supplementary Table 3) and the error tolerant search are as follows:
| Mass-tolerant (open) Search | |||
|---|---|---|---|
| Bin | Delta | Count | Assignment |
| 234 | -0.0002 | 339578 | (unmodified) |
| 252 | 15.9944 | 21171 | Oxidation |
| 277 | 43.0059 | 13660 | Carbamyl |
| 236 | 1.0259 | 12741 | 13C |
| 235 | 0.9608 | 11747 | Deamidated |
| 237 | 1.9755 | 7614 | Should be 2.01, 13C2? |
| 216 | -17.0255 | 6627 | Ammonia-loss, Gln->pyro-Glu |
| 399 | 301.9864 | 5600 | ? |
| 233 | -0.9464 | 4521 | artefact |
| 287 | 53.9190 | 3326 | Cation:Fe[II] |
| 264 | 27.9946 | 3285 | Formyl |
| 232 | -1.0281 | 3185 | artefact |
| 230 | -2.0534 | 2599 | artefact |
| 269 | 31.9893 | 2561 | Dioxidation |
| 333 | 183.0367 | 2290 | AEBS |
| 254 | 16.9961 | 2030 | Oxidation+13C? |
| 189 | -89.0305 | 1934 | Met-loss+Acetyl |
| 305 | 79.9666 | 1866 | Phospho |
| 318 | 128.0964 | 1588 | Lys |
| 231 | -1.9276 | 1573 | artefact |
| 239 | 3.0216 | 1514 | 13C3? |
| 238 | 2.9008 | 1272 | artefact |
| 369 | 249.9803 | 1254 | ? |
| 292 | 57.0227 | 1108 | Carbamidomethyl |
| Error tolerant Search | ||||
|---|---|---|---|---|
| Modification | Site | Delta | Count | Notes |
| Carbamidomethyl | C | 57.0214 | 136316 | Fixed mod in search |
| Oxidation | M | 15.9949 | 79590 | Variable mod in search |
| Non-specific cleavage | - | - | 16836 | |
| Carbamyl | N-term | 43.0058 | 13056 | |
| Gln->pyro-Glu | N-term | -17.0265 | 8094 | |
| Deamidated | N | 0.9840 | 7295 | |
| AEBS | Y | 183.0354 | 4472 | |
| Dioxidation | W | 31.9898 | 3984 | |
| Formyl | S | 27.9949 | 3761 | |
| Ammonia-loss | N-term | -17.0265 | 2919 | pyro-carbamidomethyl |
| Phospho | S | 79.9663 | 2669 | |
| AEBS | K | 183.0354 | 2529 | |
| Acetyl | N-term | 42.0106 | 2510 | |
| Formyl | T | 27.9949 | 2153 | |
| Oxidation | W | 15.9949 | 2117 | |
| Deamidated | Q | 0.9840 | 1848 | |
| Carbamyl | K | 43.0058 | 1699 | |
| Glu->Gln | E | -0.9840 | 1514 | same as amidation |
| Arg | N-term | 156.1011 | 1275 | ISD / non-specific cleavage |
| Carbamyl | T | 43.0058 | 1224 | |
| Cation:Fe[II] | D | 53.9193 | 1172 | |
| Iodo | Y | 125.8966 | 1138 | |
| Cation:Fe[II] | E | 53.9193 | 1132 | |
| Delta:H(2)C(2) | N-term | 26.01565 | 1121 | |
| Carbamyl | S | 43.0058 | 1091 | |
| Ammonia-loss | N | -17.0265 | 1030 | |
The frequency distributions for the bins illustrated in Figure 2 of the paper are narrow Gaussians, but some of the other bins with high counts extend over a very wide mass range and are not well fitted by a Gaussian, so have been labelled artefact. An example would be bin 231, which tails from -1.98 to -1.80. Bin 237 is listed with a mass of 1.9755 and a count of 7614, but is actually a broad distribution extending from 1.8 to 2.1 with a spike at 2.01.
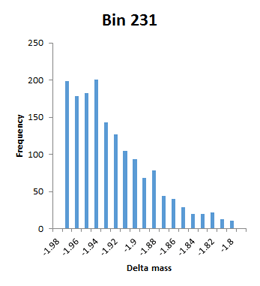
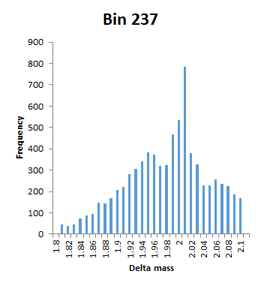
This is a concern, because the mass accuracy is good – low ppm. The channels in these histograms are 0.01 Da wide, so any particular modification can only acount for one or two channels at most. There is almost a continuum of delta mass values in certain ‘busy’ regions, and it is difficult to imagine coming up with any sort of assignment for most of them. First guess might be that these are mostly false matches, but the authors argue strongly that the peptide FDR is well below 1%. Further investigation is clearly required.
Otherwise, many of the modifications appear in both lists and are the "usual suspects". Those with a question mark are not discussed or assigned in the paper, although I’m sure the authors must have puzzled over them.
For small mass values, you might hope the mass accuracy would be sufficient to give an elemental composition. Unfortunately, these are mass differences, and the counts of some elements may be negative, (e.g. deamidation is H-1 N-1 O). There are some nice online tools to find elemental compositions from mass values, such as ChemCalc, but I haven’t found one that can handle negative counts.
SNPs are not a reasonable assignment when the matches are scattered across a large number of different sequences, so it seems unlikely that 16.9961 is Asn->Met, even though the mass is a good fit. Highly abundant delta masses that are not in Unimod are more likely to be combinations of common modifications than truly novel moieties. For an assignment to a combination to be credible, the individual modifications need to be even more abundant. On these grounds, Oxidation + 13C might be a reasonable assignment for 16.998.
As mentioned, 2.01 is a more representative mass value for bin 237 than 1.9755, in which case, we could assign it as 13C2 and maybe bin 239 is 13C3.
This leaves question marks against two of the most abundant delta masses from the open search: 249.98 and 301.99. For such large masses, there are very many possible combinations. Any good suggestions out there? This illustrates a significant drawback of the open search – if you find an abundant modification that isn’t in Unimod, how do you figure out what it is?
Among the less abundant modifications, all those discussed in the paper are in Unimod except an unidentified 72.005 Da modification to N-terminal tryptophan, an unidentified 103.063 Da cysteine modification, and some polyalanine insertions found in ribosomal protein L14. Diphthamide (called diphthalamide in the paper) was in Unimod but with one too many hydrogens, (since corrected).
The authors describe how matches in an open search are weaker because only unmodified fragments are used for the match. For spectra that have strong b and y ions, this isn’t a huge problem. For spectra that are mostly y ions, there is a bias against modifications towards the C-terminus, because this takes out most of the potential fragment peak matches. For spectra that are mostly b ions, the bias is against modifications towards the N-terminus. Also, the search engine cannot make use of known neutral loss behaviour, such as loss of 98 from phosphate. On the other hand, modifications that are lost in their entirety on fragmentation, such as glycosylation or sulfation, so that fragments revert to their unmodified masses, should give matches that are just as strong as in a standard search. (Although sulfate is not one of the modifications identified in the open search.)
In an error tolerant search, although the search is limited to peptides with a single unsuspected modification, the matches are just as strong as if the modification had been specified as a variable modification in a standard search. Modified fragments can be matched and neutral loss information applied.
On balance, it is difficult see the open search becoming widely used for shotgun proteomics because it requires so much more time and effort for results interpretation compared with a multi-pass search. On the other hand, an important potential application of the open search is not mentioned anywhere in the paper – characterising modifications on endogenous peptides. Multi-pass searches are of limited use in this case because a protein will often be represented by a single peptide. If that peptide is modified, it is very likely to be missed. The open search may provide a more efficient alternative to the current strategy of de novo followed by an error tolerant sequence tag search.
Keywords: error tolerant, mass tolerant, modification, open search, Unimod
Maybe UniMod can be updated to encompass experimentally-discovered open mods such as 72.005, 103.063, etc., annotated, at least temporarily, as “Unknown” with a reference.
That way, error-tolerant approach can never be a substantially worse approach.
Maybe it is a complementar search method different to direct search for modification.