Generating high quality spectral libraries for DIA-MS
Using Mascot Daemon, Mascot Distiller and Mascot Server
A recent paper from Manda et al.[1] describes a pipeline to generate high quality spectral libraries from Data-Dependent Acquisition (DDA) experiments for use in searching Data-Independent Acquisition (DIA) data, starting from the raw data, through to generating the library from the search results. The pipeline uses a collection of different tools in order to generate a library suitable for DIA data analysis.
We introduced the ability to search spectral libraries in Mascot Server 2.6, using the NIST MSPepSearch spectral library search engine. As part of this, we included the ability to generate your own spectral libraries from your Mascot search results. The generated libraries are in the NIST MSP format, which several of the DIA search tools, such as DIA-NN and Skyline, can also use.
In this blog, we’ll use the dataset used by Manda et al. to generate MSP format spectral libraries using Mascot Distiller and Mascot Daemon to process and search the raw data files, and then the spectral library crawler in Mascot Server to generate the library files. You can find the fractionated HEK293 cell line dataset available for download here (9.3GB)
One advantage of using Mascot to do this is if you’re using ETD fragmentation on your instrument. Although the MSP format only supports a-, b- and y- ions, Mascot will export c-, x- and z- ions as b- and y- ions with a suitable neutral loss. For a DIA search, another advantage of using a spectral library created from DDA search results over one created by in-silico prediction is the ability to include a range of variable modifications not included in the in-silico prediction tool’s model.
Step 1: Determine suitable search parameters for the dataset
Mascot was one of the search engines used to generate results by Manda et al., so suitable search settings are available in the supplementary data for the paper. However, in order to fully outline the process, we’ll use the procedure from this blog article. We can use an error-tolerant search in Mascot in order to find the most abundant modifications and use these in the searches from which we’ll create the library. To do this, we first selected one of the raw fraction files from the dataset and processed it using Mascot Distiller. We then carried out an integrated Error Tolerant search against the UniProt Human proteome with integrated decoy using Mascot Server 2.8 using the following search settings:
| Target FDR | 1% |
| Enzyme | Trypsin |
| Fixed modifications | Carbamidomethyl (C) |
| Peptide mass tolerance | 25 ppm |
| Fragment mass tolerance | 0.05 Da |
So, we’ve included the fixed modification for alkylation, but left the variable modifications clear. When completed, we can look at the search result header to find the most abundant mods – these are the ones we need to specify as fixed or variable mods when we do the searches to create the library:
| Modification | Delta | Type | Site | Total matches |
|---|---|---|---|---|
| Carbamidomethyl | 57.021464 | fixed | C | 2419 |
| Non-specific cleavage | ET | - | 351 | |
| Guanidinyl | 42.021792 | ET | N-term | 139 |
| Carbamidomethyl | 57.021464 | ET | M | 118 |
| Deamidated | 0.984016 | ET | N | 110 |
| Carbamidomethyl | 57.021464 | ET | N-term | 74 |
| Oxidation | 15.994915 | ET | M | 55 |
Some interpretation of the table is required. Most of the assignments look reasonable, but Guanidinyl doesn’t seem the most likely assignment for the delta of ~42Da. The Error Tolerant search suggests a number of possible modification assignments for a given mass-delta and position, but the table (for reasons of legibility) only reports the first in the list – the important piece of information is actually the Delta. Much like the plus one dilemma, we have a +42 dilemma, as there are several possible assignments all within a very close mass range around +42Da which may explain the observed shift – for example, protein N-terminal acetylation. If we take a closer look at some of the matches, we can quickly see that this is indeed the most likely assignment, as we find the majority of matches at the protein N-terminus as shown below:
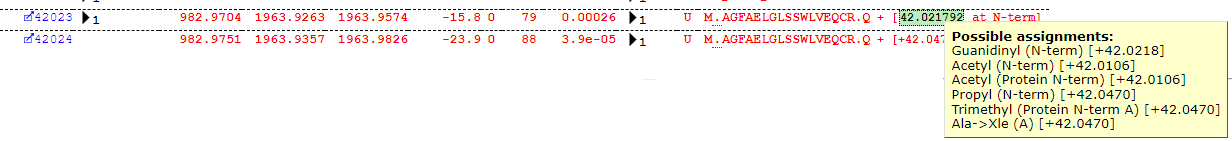
In addition to the identified modifications, there are a large number of non-specific cleavage matches identified by the error tolerant search pass, which were not addressed in the original paper, so we’ll capture these in our searches as well.
Step 2: Perform standard searches for abundant modifications and non-specific cleavage products
On the basis of the above, we’ll perform two sets of searches, one for fully tryptic peptides and a follow-up search for any peaklists which don’t get a significant match with semiTrypsin. This allows us to keep the search space smaller for the initial search, then try to find additional semiTryptic peptides in a separate search. In both cases we’ll search against a contaminants and the Uniprot human proteome databases, with Deaminated (NQ), Acetyl (Protein N-term), Carbamidomethyl (M), Carbamidomethyl (N-term), Oxidation (M) as variable modifications, automatic decoy search selected with a target FDR of 1%. We can do this in Mascot Daemon by first setting up a follow-up search with semiTrypsin selected as the enzyme, then set up a normal batch task. When creating the batch task, we specified that any peptides with an expect value of less than 0.01 be passed to the follow-up task to be researched using semi-trypsin specificity. In both cases, we used Mascot Distiller to automate peak-detection on the raw files. Using Daemon like this allows us to setup common search titles for the two groups of searches, which will be useful when selecting the correct searches to generate the spectral libraries later on in the process.
When running the searches remember that Mascot will only match 1+ and 2+ ions series, so if you have higher charge state fragment ions present, these won’t be annotated in the library. If you want to decharge the peaklists in order to improve coverage then the generated library will also contain only decharged fragment ions, which you will need to take into account when running any searches against the library, either in Mascot or when using the library with an external tool such as Skyline or DIA-NN.
Step 3: Calculate an expect value cut-off for approximately 1% peptide FDR for each search set
We want to include only reliable peptide matches in our spectral libraries, so we want to specify a threshold when we create the libraries which will give us approximately 1% FDR. The easiest way to do this is to open a merged result set from Mascot Daemon, then use the settings to adjust the significance threshold to give us a 1% PSM FDR for the fully- and semi- trypsin search results:
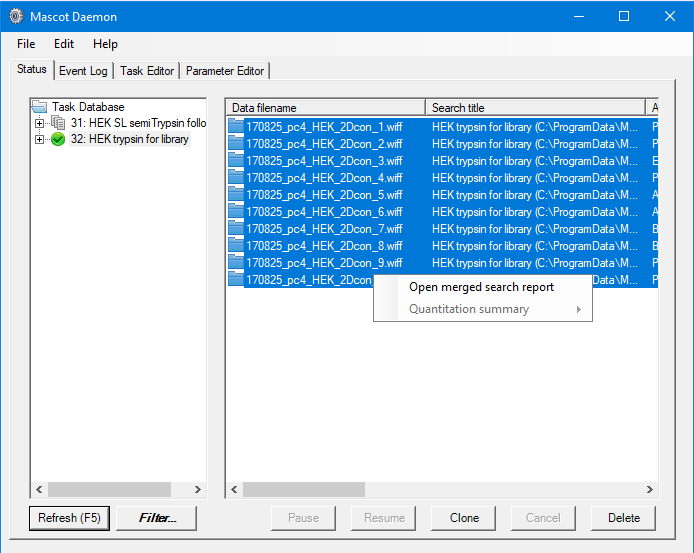
Because we used Mascot 2.8 and set the target FDR in the search settings, the merged results actually opened at the target FDR. Using this we have the following thresholds:
- Trypsin search threshold: 0.00679
- semiTrypsin search threshold: 0.001721
Step 4: Create the libraries
Because the thresholds for the two sets of searches are so different, we’ll create two separate spectral libraries for this dataset – one for the Trypsin search results, and a second library for the semiTrypsin searches.
To create a new library in Database Manager, choose Library; Create new; Create from search results. Work your way through the setup Wizard – in most cases the default values will be acceptable, though you’ll need to select a suitable reference database. When you’ve done that, choose Edit filters. We only want reliable matches in the library, and we only want to include matches from the trypsin searches in one library, and the semi-trypsin follow-up searches in the second. We can filter on the enzyme, database and peptide expect score, which allows us to separate the results in order to create our two libraries and exclude matches to the contaminants database:
Filter settings for HEK_Trypsin library: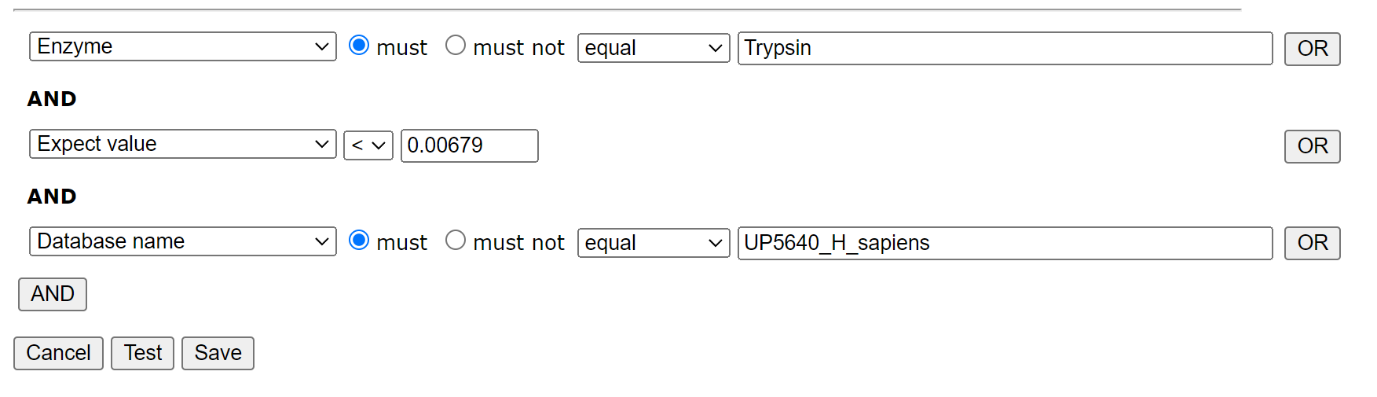
Filter settings for HEK_semiTrypsin library: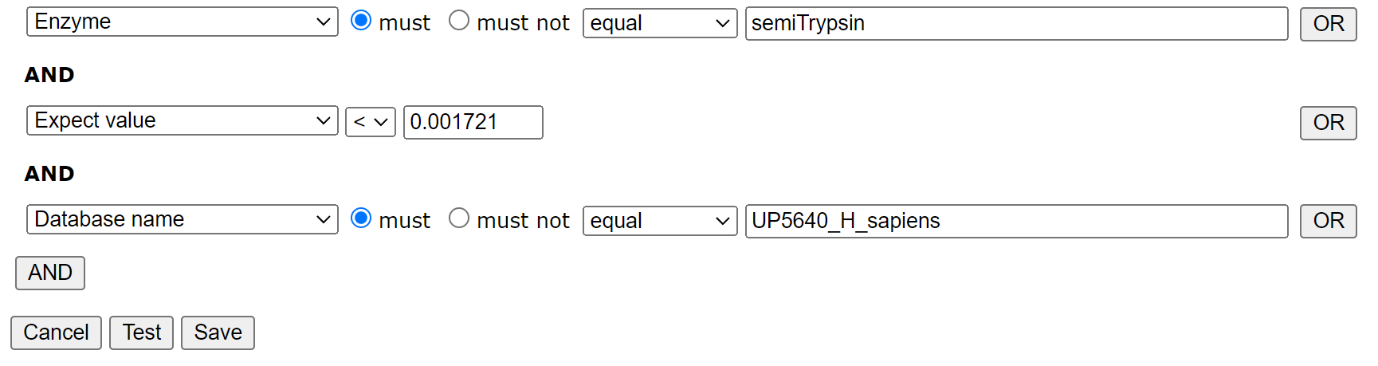
Choose Import search results and set the filters to ensure that only the result files created in step 3 are scanned. In some cases, it will be simplest to copy the result files to a new folder, so that they can all be selected in one go by a simple wild card path. However, in this case we defined the search titles in a way which differentiated between the trypsin and semiTrypsin searches in step 2, so we can filter on the search date and titles.
5. Use the libraries
The libraries will now be available for use in Mascot and you’ll find the MSP file under the sequence directory on the Mascot server:
You can now copy the MSP from the library’s ‘current’ directory and use it with dedicated DIA tools such as Skyline and DIA-NN.
In addition, when the library is created, a full import log is available in Database Manager documenting exactly what was and was not imported into the library. These log files are kept in the library’s ‘incoming’ directory on the filesystem.
Reference
1 Srikanth S. Manda, Zainab Noor, Peter G. Hains, and Qing Zhong, PIONEER: Pipeline for Generating High-Quality Spectral Libraries for DIA-MS Data. Current Protocols, 1, e69. doi: 10.1002/cpz1.69
Keywords: DIA, Mascot Daemon, Mascot Distiller, Mascot Server, spectral library