Quantitation: emPAI protocol
The Exponentially Modified Protein Abundance Index (emPAI) offers approximate, label-free, relative quantitation of the proteins in a mixture based on protein coverage by the peptide matches in a database search result. Developed by Ishihama and colleagues, the key publication is Ishihama, Y., et al., Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein, Molecular & Cellular Proteomics 4 1265-1272 (2005)
Unlike the other quantitation protocols, the information required for emPAI is always present in a search result, and there are no parameter settings, so emPAI is "always on", as long as the MS/MS search contains at least 100 spectra.
The formula is very simple: 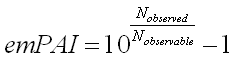
Where Nobserved is the number of experimentally observed peptides and Nobservable is the calculated number of observable peptides for each protein. The tricky bit is deciding what to include and what to exclude in these two counts.
The number of observed peptides
The count of observed peptides only includes peptide matches with scores at or above the homology threshold, or the identity threshold, if there is no homology threshold. Ishihama et. al. obtained best proportionality for a standard protein mixture by counting unique parent ions, including different charge states from the same peptide sequence. Mascot 2.4 and earlier followed this same rule, which works well for singly and doubly charged data. However, if peptide matches exist in a number of charge states, such as 2+, 3+, 4+, 5+ and 6+, the rule causes emPAI to be overestimated. Mascot 2.5 and later count unique parent ions only once, regardless of charge state. The difference from the original rule for singly and doubly charged data is negligible compared to the other sources of certainty, as described below.
The number of observable peptides
To estimate the number of observable peptides, Ishihama et. al. performed explicit in silico digests of the protein sequences. The peptide list was then filtered to exclude peptides outside the mass spectrometer scan range and the observed nano-LC retention time range.
For reasons of speed, we prefer to make a calculated estimate of the number of observable peptides based on the protein mass, the average amino acid composition of the database, and the enzyme specificity. The error of doing this is negligible compared with other sources of uncertainty:
- It isn’t practical to filter by retention time, because this information is usually unavailable
- The mass range of the instrument has to be estimated from the range of precursors found in the data set
- Mass range filtering is by Mr, rather than m/z
- The digest is assumed to be a limit digest
- No obvious way to extend the calculation to semi-specific or non-specific digests
In the supplementary material for Ishihama et. al., there is a worked example for human serum albumin which resulted in a count of 34 for the observable peptides in the Mr range 700 to 2800 and the retention time range 40 to 150 minutes. The enzyme was strict trypsin and no missed cleavages were allowed. The number of peptides estimated by the routine used here is 35.
Example
Click here for an example of emPAI. We are grateful to Dr Jyoti Choudhary of the Sanger Institute for this small, LC-MS/MS data set from a human cell lysate acquired using a Waters QTof. Notice how hit 3 has a larger emPAI value than hits 1 or 2, even though it has fewer matches. This is because TRY1_BOVIN is less than half the size of the other proteins, so will have a lower Nobservable.
Reasons for missing or meaningless emPAI values:
- No emPAI values if fewer than 100 queries in the search
- Meaningless emPAI values if semi-specific enzyme or no enzyme
- No emPAI values if old-style (manual) error tolerant search
- For an integrated decoy search, no emPAI values in the report for hits in the decoy database.
- emPAI values are only reported for the primary protein in the hit, not for same set or sub-set proteins.
- An emPAI value may be missing if the protein was a very weak hit and the protein mass was not saved to the result file and the sequence database is no longer on-line or the protein has disappeared from the sequence database. In such cases, the protein description and mass will also be missing from the report.

