Quantitation: Configuration
Both Mascot Server and Mascot Distiller use the same XML configuration file to define quantitation methods. This file, called quantitation.xml, lives on the Mascot Server and is downloaded by Mascot Distiller and other clients as required. The XML schema can enforce some constraints, but many elements and attributes are just labels for logic embedded in code.
Configuration editor
Provided you have the necessary security rights, the Mascot configuration file editor can be accessed by following a link from your local Mascot home page. Choose Quantitation to edit, copy, or delete existing methods and create new ones. Choosing print displays a printer friendly summary of the quantitation method.
For each method, a tabbed dialog is used to navigate between property pages. In many cases, the property pages correspond to XML elements, but sometimes elements have been combined onto a single page or split across multiple pages so as to give a balanced layout.
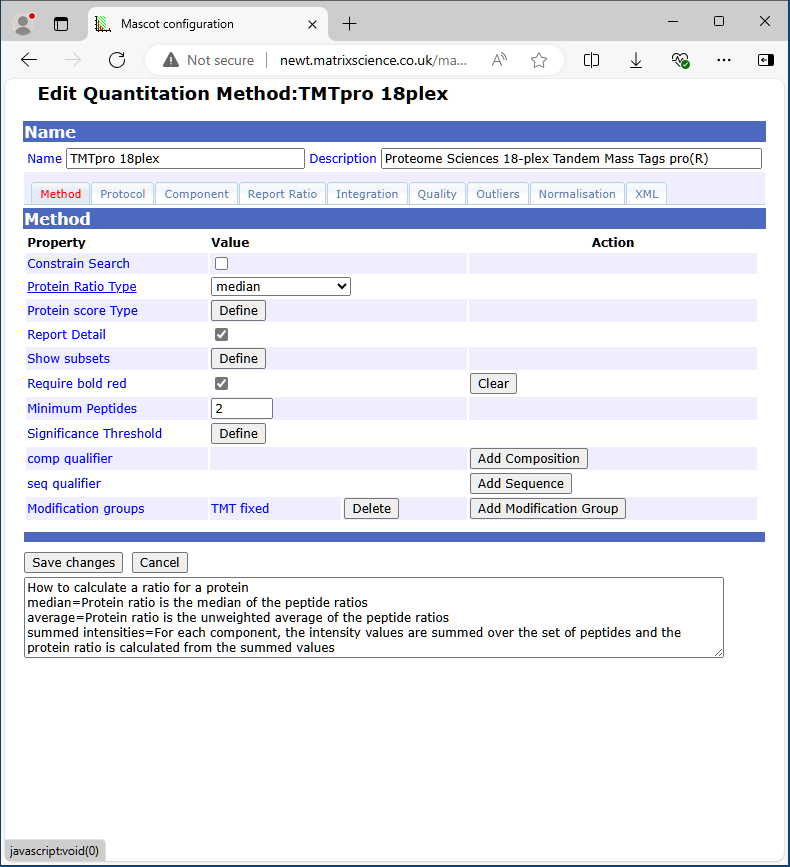
When the mouse pointer moves over a field label, help text is displayed in the text area at the bottom of the window.
Nothing is saved to the configuration file until you choose the Save changes button. The Cancel button discards any changes and returns you to the list of quantitation methods.
Acknowledgement: The tabs in the configuration editor use CSS sliding doors as refined by explodingboy.
Reference
Each method has a Name, which is required and will appear in the quantitation drop down list on the search form, and an optional, free-text Description, which will be displayed in the report header. In the XML file, these are attributes of the method element, but in the configuration editor, they appear above the tabbed property pages, so as to be visible whenever a method is being edited.
In this reference section, the names are those that are displayed in the configuration editor. The name of the corresponding element or attribute in the XML file may be different. Where appropriate, this name will be given in green, together with details of whether a value is required or optional and any default.
Method
Constrain search (constrain_search, optional, default true)
In many quantitation experiments, separate samples are derivatised then pooled. Thus, a given peptide may carry one or another set of modifications, but never a mixture. This is specified by creating a group of exclusive modifications, which can be thought of as a choice of fixed modifications.
If constrain search is true, then this constraint is applied during the search, preventing any matches to peptides with mixed or partial labelling. This will have the advantage of speeding up the search and reducing the significance thresholds, but the disadvantage of hiding evidence of experimental defects leading to incomplete labelling. If constrain search is false, the Mascot search will treat exclusive modifications as variable, and the tighter constraint, of treating them as a choice of fixed modifications, will only be applied when generating the quantitation report.
Protein ratio type (protein_ratio_type, optional, default average)
The way a protein ratio is calculated from a set of peptide match ratios.
- median: The median peptide ratio is selected to represent the protein ratio. If there are an even number of peptide ratios, the geometric mean of the median pair is used
- average: The protein ratio is the geometric average of the peptide ratios
- Summed intensities: For each component, the intensity values are summed over the set of peptides and the protein ratio(s) calculated from the summed values. This will be the best measure if the accuracy is limited by counting statistics. This is only available for reporter protocol and, when selected, SD(geo) and p-values for the protein ratios are not available.
Protein score type (prot_score_type, optional)
Corresponds to the Mascot report format parameter. Provided here to enable a quantitation method to control the initial appearance of the summary report, over-riding default behaviour or mascot.dat settings.
Report detail (report_detail, optional, default true)
Whether to display quantitation data at the peptide match level
Show subsets (show_sub_sets, optional)
Corresponds to the Mascot report format parameter. Provided here to enable a quantitation method to control the initial appearance of the summary report, over-riding default behaviour or mascot.dat settings.
Require bold red (require_bold_red, optional)
Corresponds to the Mascot report format parameter. Provided here to enable a quantitation method to control the initial appearance of the summary report, over-riding default behaviour or mascot.dat settings.
Minimum peptides (min_num_peptides, optional, default 2)
A ratio for a protein hit is only reported if this minimum number of assigned peptide matches have valid ratios.
Significance threshold (sig_threshold_value, optional)
Corresponds to the Mascot report format parameter. Provided here to enable a quantitation method to control the initial appearance of the summary report, over-riding default behaviour or mascot.dat settings.
Comp qualifier (comp element, optional)
An amino acid composition qualifier that must be matched for a peptide to be used for quantitation. For example, *[C] would be used for ICAT. Only a single comp qualifier is supported.
Seq qualifier (seq element, optional)
A sequence qualifier that must be matched for a peptide to be used for quantitation. Choose the Add Sequence button to add a new seq qualifier field and choose the adjacent Delete button to remove one.
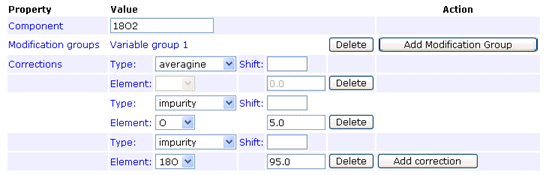
Modification groups (modifications element, optional)
Method level modification groups, which may be fixed or variable, can be added, edited, or deleted. Choosing to add or edit a modification group displays the Modification group property page …
Modification Group
In a quantitation method, modifications are organised into groups and classified as fixed, variable, or exclusive. At the component level, modification groups can be variable or exclusive, and serve to identify the component. They can also be defined at the method level, but only as fixed or variable. Defining modifications at the method level is a convenience, and saves having to choose them in the search form. A modification group can also contain a declaration that residue(s) or a terminus are unmodified.
The following rules apply to modification groups:
- Any particular modification can only appear once in a given method
- If exclusive groups are defined, then all peptides used for quantitation must fit to one of these groups.
- Each specificity that appears in an exclusive group is no longer available for use elsewhere. For example, if an ICAT method uses exclusive groups for the ICAT modifications, it would be illegal to specify any other modification for C in a fixed or variable modifications group or in the search form.
- At the component level, only exclusive and variable modification are allowed, with a maximum of one exclusive group per component.
- At the method level, only fixed and variable mods are allowed.
- Exclusive groups are effectively a choice of fixed modifications, so the restrictions that apply to fixed modifications also apply. That is, you cannot have multiple neutral losses, peptide neutral losses, "residue@terminus" specificity or protein terminus specificity in an exclusive (or fixed) group.
- Component level isotope elements do not apply to modifications defined at the method level, or in the search form or MGF file.
- If a component has both modifications and isotope elements, the isotope element applies to the component level modifications.
Name (name, optional)
A modification group only requires a name if you intend to refer to it from an exclusion element
Mode (mode, required)
The mode can be fixed, variable, or exclusive. An exclusive group is only allowed at the component level whilst a fixed group is only allowed at the method level. All of the exclusive groups form a closed set, which effectively represents a choice of fixed modifications. This may be a simple pair, such as one component modified and the other unmodified, or a triplet, e.g. ICPL, ICPL:2H(4), and ICPL:13C(6). Hence, there will usually be either no exclusive groups, or at least two. A single exclusive group has no value and is probably an error.
Required (required, optional, default false)
If required is set true for a modification group, a peptide will only be quantified if the modification group is present in the peptide. The main occasion when this comes in useful is for reporter protocol when a tag such as iTRAQ or TMT has been used to label the protein prior to digestion with trypsin. This labels the protein N-terminus and all Lysine residues, which means that roughly half the peptides are unlabelled and need to be excluded from quantitation by checking this option.
Modification (mod_file, optional)
A Mascot modification chosen from a drop down list.
Un modified (unmodified, optional)
A declaration that residue(s) or a terminus are unmodified. This is defined, Unimod-style, as a combination of a site and a position. The site can be a residue or a terminus. The position is a choice of Anywhere, Any N-term, Any C-term, Protein N-term, or Protein C-term. Protein terminus positions are not advisable for component level modifications because there is no guarantee that the experimental protein terminus is the same as the end of the database sequence. Certain combinations of site and position, like N-term at Any C-term, are clearly illegal. If the site is a terminus then Anywhere is synonymous with Any N-term or Any C-term.
Local definitions (local_definition, optional)
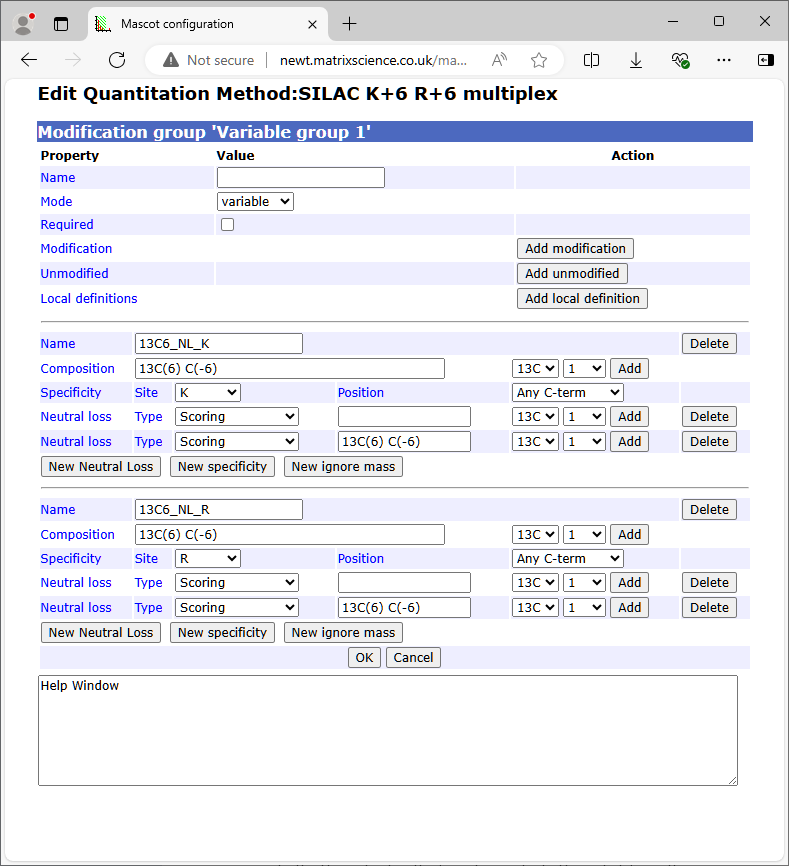
A modification can be defined within the quantitation method if it is not available from Unimod or would not be appropriate for Unimod. If the name of a local definition already exists in Unimod, then the local definition is used, and masks the Unimod entry. The format is similar to Unimod, defining the modification in terms of a difference in elemental composition. Compositions can be typed into the edit field or added by selecting values from the adjacent drop down lists.
A modification specificity is defined by a combination of site and position, together with Neutral losses of various kinds. You can have multiple specificities, but only if they are all "residue@anywhere". If any specificity is a terminus, or at a terminus, then that must be the only specificity.
For a more complete description of how to define a modification, refer to the relevant Unimod help pages. For more about neutral losses, refer to the Mascot modifications help page.
Protocol
The protocol tab contains a drop down list of the supported protocols. According to the selection, it may also display controls for some protocol dependent parameters. Details of these can be found on the individual protocol help pages:
Component
The component tab describes how to identify a single component of a ratio to be reported, together with any component specific parameters. A component must have a name, and this name must be unique within the method.
Depending on the protocol, the component tab will include controls for one or more of the following categories:
Protocol Reporter Precursor Multiplex Replicate Reporter ion m/z X Modification group X X Isotope X File index X Correction X X X The component tab has a drop down list for any existing components, together with new, copy, and delete buttons. There will also be an Add button to add controls for each category from the table above which applies to the selected protocol. These are:
Reporter ion m/z
monoisotopic (required), average (required)
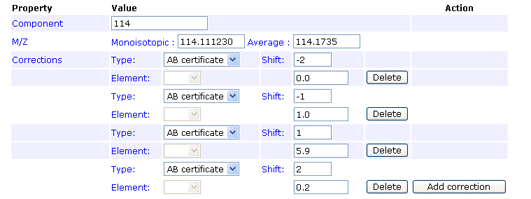
The monoisotopic and average m/z values of a single reporter ion peak. For example, in iTRAQ, one of the peaks used for quantitation has a monoisotopic m/z of 114.111230 and an average m/z of 114.1735
Modification groups
Component level modification groups, which may be variable or exclusive, can be added, edited, or deleted. Choosing to add or edit a modification group displays the Modification group property page, described above.
Isotopes
old, new
This is the isotope substitution that identifies a component in a metabolic labelling experiment. Most likely, the light component will be N, (i.e. 14N), and the heavy component 15N. Any elements which are not specified default to their most abundant natural isotope. Hence, there is no need to explicitly specify C, H, etc., for the heavy component in a 15N experiment. The light component can be specified as having N for both old and new, or left "empty".
Note that changing the mass of an element only changes the residue masses and any variable modifications defined at the component level. Modifications defined at the method or search form level are not shifted because they are assumed to be artefacts, which will not contain the metabolic label.
File index (file_index, optional)
File index is a zero based integer that identifies a data set for label-free quantitation using the Replicate protocol. This is only important in respect of how ratios will be reported. Hypothetically, if there were 4 files called A, B, C, and D, you might want to report B/A and D/C or B/A, C/A, and D/A, etc.
Isotope corrections may be necessary because of label impurity. For example, a batch of 18O water might be 95% 18O and 5% 16O. Also, when the peaks used for quantitation are very close together, a correction may be required because the isotopic envelopes of the components have a significant overlap.
Mascot quantitation supports three correction types: AB certificate, averagine, and impurity. AB certificate handles the corrections for reporter ion peaks, such as iTRAQ, and cannot be combined with the other two types. The averagine type handles isotopic envelope overlap. The impurity type is for label impurity.
If the protocol is precursor, an averagine correction is always applied, whether or not specified in the method. If the protocol is reporter or multiplex, be careful that there is no conflict between these corrections and any prior de-isotoping of the peak list. If the fragment ions have been accurately de-isotoped, then no further corrections are required. On the other hand, it is extremely difficult to de-isotope peaks that may or may not contain isotope labels. Simple de-isotoping routines that select the most intense peaks and then delete any satellite peaks 1 or 2 Da higher in mass will do more harm than good.
Type
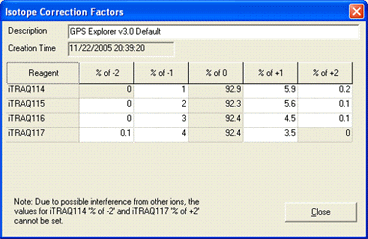
AB certificate makes it easy to enter isotope impurity values directly off the certificate of analysis supplied with a batch of AB SCIEX iTRAQ reagent. Each component corresponds to a row on the certificate, and the correction is entered as pairs of mass shift and percentage values. For example, if the certificate looked like this:
The first row would be entered like this:
You can enter the 0 Da shift of 92.9% if you wish. If not, a value will be assigned automatically to bring the total for the row to 100%. Note that, if you enter a value for the zero shift, the row must add up to exactly 100%, or an error will be reported.
When averagine is selected, the isotope envelope at any particular mass is calculated by assuming the peptide to be composed of averagine, the "average" amino acid. If this type of correction is specified for one component in a method, it will automatically apply to all components. The approach is similar to that described in Johnson, K. L. and Muddiman, D. C., A method for calculating 16O/18O peptide ion ratios for the relative quantification of proteomes, JASMS 15 437-445 (2004).
The impurity correction type handles reagent impurity, such as this example for 18O labelling where the heavy water is 95% pure:
In any given method, an impurity correction can only be entered for isotopes of a single element. Don’t try to mix isotopes from different elements. The correction factors only need to be entered under one component. Entering duplicate corrections for all components is not an error, as long as they are all identical, but serves no purpose.
Footnote: There is an implicit assumption here that the impurity correction only applies to the heavy isotope(s), and works "downwards". That is, in the example above, some of the intensity of peptides labelled with the 18O label will appear at lower mass values. There is no mechanism to specify that the "unlabelled" peptides contain (say) 5% 18O. The averagine correction is an "upward" correction, but it is always based on the natural abundances of heavy isotopes. The AB certificate type combines both upward and downward corrections for the general case in which there are isotopes from multiple elements and nothing can be assumed to be "natural". The correction factors are obtained experimentally, by analysing the isolated reagents. If you need to do something similar, which is not supported by the averagine or impurity types, the AB certificate type provides a possible route.
Report ratio
Ratio
Identifies a ratio to be calculated and reported. The text must be unique and will be used to label the ratio in any report. For example, 115/114 or heavy / light.
Numerator, Denominator, Coefficient
Both numerator and denominator can be linear combinations. In an 18O experiment, for example, you might want to report the ratio of (18O1 + 18O2) / 18O0.
In the XML file, each ratio is defined using a report_ratio element, with the label text as the name attribute. Each report_ratio element has at least one child numerator_component element and one child denominator_component element, which specify the component name and a coefficient as attributes.
Integration
These settings control how signals are extracted from the raw data file(s) by Mascot Distiller. In most cases, this will be integration of peaks across a range of survey scans.
Integration method (method, required)
How the data are integrated
- none: No integration
- simpsons: Simpson’s rule
- trapezium: Trapezium rule
If Simple Ratio is checked, you must choose either simpsons or trapezium, and the XIC peak will be integrated using the specified method. If simpsons is chosen, but the time intervals between the source scans in the XIC peak are not constant to +/- 1%, trapezium will be used instead. Ratios are calculated using the XIC peak areas.
When Simple Ratio is not checked, which is the default, the reported ratios are determined by fitting a straight line through intensity points for the scans in the XIC peak range, as described here, and Integration Method setting is ignored.
Integration source (source, optional, default survey)
What is integrated
- survey: precursor intensity from survey scan
- zoom: precursor intensity from zoom scan
For standard trap data with zoom scans, zoom will usually give the best results
Mass delta (mass_delta, optional), (mass_delta_unit, optional, default Da)
The mass tolerance for pairing up components in replicate protocol.
Allow Elution Shift (allow_elution_shift, optional, default false), (elution_time_delta_unit, optional, default seconds)
Whether to try and time align components. Should always be true for replicate protocol. Should be false for Precursor protocol unless one of the isotope labels contains deuterium.
Elution time delta (elution_time_delta, optional), (elution_time_delta_unit, optional, default seconds)
The time tolerance for pairing up components in replicate protocol.
Elution profile correlation threshold (elution_profile_correlation_threshold, optional)
Threshold on the standard error for a straight line fit of the component intensities from each of the scans in the XIC peak. (Nothing to do with elution time shift.)
All charge states (all_charge_states, optional, false)
All charge states extends the mass and time matching to cover contiguous charge states. In other words, if there is a positive match on 2+, the software will look for 1+ and 3+. It will not look for 4+ unless 3+ is found, etc.
All charge states threshold (all_charge_states_threshold, optional, default 0.2)
When all_charge_states is true, additional contiguous charge states are included in quantitation if their intensity exceeds this fraction of the most intense charge state for which there is a database match.
Matched Rho (matched_rho, optional, default 0.7)
Threshold on the correlation coefficient between the predicted and observed precursor isotope distributions.
XIC threshold (xic_threshold, optional, default 0.1)
The start and end of an XIC peak are where the intensity drops to this fraction of the intensity of the XIC peak maximum.
XIC max width (xic_max_width, optional, default 250)
Upper limit on the number of survey scans in an XIC peak
XIC smoothing (xic_smoothing, optional, default 3)
XIC peak is smoothed by a set of 2n+1 Savitzky-Golay convolution integers, where n is this value, (0 corresponds to no smoothing).
Quality
Miscellaneous quality criteria that peptide matches must meet before they can be used for quantitation
Minimum precursor charge (min_precursor_charge, optional, default 1)
Unsigned integer, corresponding to abs(charge), in case anyone is using negative ions
Isolated precursor (isolated_precursor, optional, default false)
If set true, filter matches using the Isolated Precursor Threshold. This can be used to reject matches where the precursor region in the survey scan contains interfering peaks.
Isolated Precursor Threshold (isolated_precursor_threshold, optional, default 0.5)
Threshold on the fraction of the peak area in the precursor region accounted for by the components.
Minimum a(1) (minimum_a1, optional, default 0.0)
Used by Dimethyl labelling. Requires the a1 peak to be present and at least the specified fraction of the base peak intensity. For example, set this to 0.1 to reject any match where the a1 peak is weaker than 10% of the base peak intensity in the MS/MS spectrum. See Hsu, J. L., et al., Beyond quantitative proteomics: Signal enhancement of the a(1) ion as a mass tag for peptide sequencing using dimethyl labeling, Journal of Proteome Research 4 101-108 (2005)
Peptide Threshold Type (pep_threshold_type, optional, default maximum expect)
Peptide Threshold Value (pep_threshold_value, optional, default 0.05)Threshold for selecting peptide matches to be used for quantitation. Very important to select good matches and exclude weak, possibly incorrect, matches. Peptide Threshold Value is only used if Peptide Threshold Type is minimum score or maximum expect
Unique Pepseq (unique_pepseq, optional, default false)
If true, only report ratios for peptide sequences that are unique to one protein hit (which may be a family containing multiple proteins).
Exclusion (exclusion element, optional)
If a peptide match contains a modification from a group specified in an exclusion element, it will not be used for quantitation. In iTRAQ, for example, peptides with modified tyrosine are excluded because the reaction is slow and so liable to be incomplete, leading to skewed ratios. Choose the Add Exclusion button to add a new exclusion drop down list and choose the adjacent Delete button to remove one. Modification groups are defined by accessing the modification groups property page from either the method or component tabs.
Outliers
When ratios for individual peptide matches are combined into ratios for protein hits, a variety of procedures are available for detecting and rejecting outliers. Full details of each outlier method can be found on the statistics help page. In the XML file, an outlier element is optional. If it is missing or if it is present and the method attribute set to none, then no outlier detection is performed
- auto: Dixon’s method will be used if the number of values is between 4 and 25, while Rosner’s method will be used if the number of values is greater than 25.
- dixons: Dixon’s r11 test, also referred to as N9, is used to detect and remove a single outlier at a time from either the upper or lower extreme of the range. The test is applicable to between 4 and 100 values. Each time a value is removed, the test is repeated.
- grubbs: Grubbs’ method is used to detect and remove a single outlier at a time from either the upper or lower extreme of the range. The test is applicable to between 3 and 100 values. Each time a value is removed, the test is repeated.
- rosners: Rosner’s method will detect and remove multiple outliers in a single pass. The test will remove up to 10 outliers from a sample of at least 25 values.
Normalisation
Whatever the quantitation method, it can be difficult to ensure that each component is treated identically at all stages in the sample work-up. If it is reasonable to expect that only a minority of the proteins in the sample will be up or down regulated, global normalisation can be applied so as to make the average or median ratios across the entire data set unity. Alternatively, known amounts of exogenous protein or peptide can be spiked into the samples and specified as the basis for normalisation.
There are three normalisation methods, or four if you include none. In the XML file, a normalisation element is optional. If it is missing or if it is present and the method attribute set to none, then no normalisation is performed
- average: For each peptide ratio, a correction factor is applied such that the geometric mean of the ratios for all peptide matches that pass the quality tests is unity
- median: For each peptide ratio, a correction factor is applied such that the median of the ratios for all peptide matches that pass the quality tests is unity
- sum: (Reporter protocol only) A correction factor is applied such that the sum of the intensities for a reporter ion peak over all peptide matches that pass the quality tests is the same for all the reporter ions
Normalisation Peptides (peptides element, optional)
Instead of all peptides, normalise on the specified peptide sequence(s)
Normalisation Proteins (proteins element, optional)
Instead of all peptides, normalise on the peptide(s) assigned to the specified Protein accession(s)
XML
The raw XML for the selected method is displayed in an edit field. You can use this to copy the method to a different Mascot Server. For example, you might copy the XML from the edit field, paste it into an email, and send to a colleague. Your colleague could then create a new quantitation method, go to the XML tab, copy the XML from the email and paste it into the edit field, then choose Update method. To save the new method, choose Save changes.
Precise details for individual data items, such as the data type and whether it is optional, can be found in the XML schema. Schema documentation has been auto-generated using xs3p.
Since a picture is worth a thousand words, here’s a diagram of the schema.
Follow this link for some general XML Schema considerations.

