Quantitation: Report format
Format controls quick reference
When the quantitation method is either Reporter or Multiplex, additional formatting controls are displayed near the top of the Mascot result report.
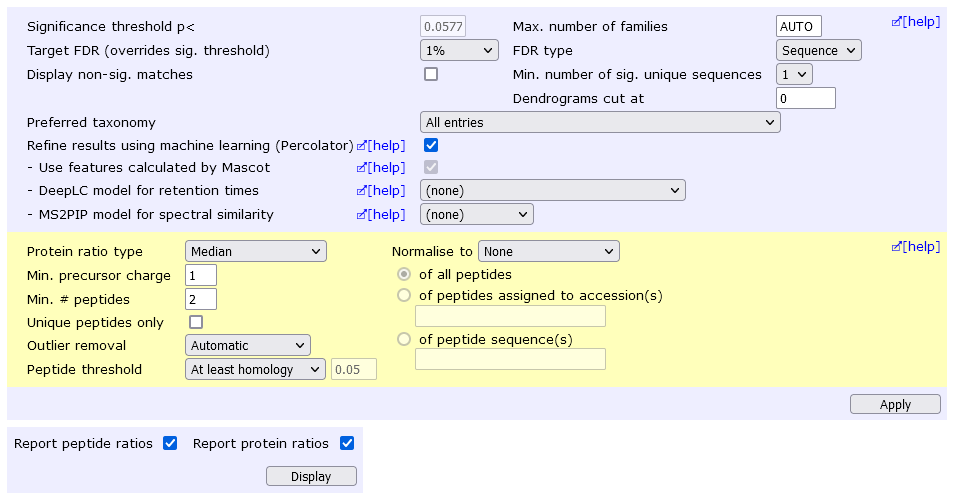
This screen shot shows the layout in the Protein Family Summary report. After making changes to the search result controls in the blue block, choose Filter to reload the report using the new settings. After making changes to the quantitation controls in the yellow block, choose Quantitate to reload the report using the new settings.
Protein ratio type: A drop down list with the choices Average, Median, and ratio of summed intensities.
- Average: The protein ratio is the geometric mean of the peptide ratios
- Median: The median peptide ratio is selected to represent the protein ratio. If there are an even number of peptide ratios, the geometric mean of the median pair is used
- Summed intensities: For each component, the intensity values are summed over the set of peptides and the protein ratio(s) calculated from the summed values. This will be the best measure if the accuracy is limited by counting statistics. This is only available for reporter protocol and, when selected, SD(geo) and p-values for the protein ratios are not available.
Min. precursor charge: A peptide ratio will not be reported if the precursor charge is less than this value. Note that this number is compared with the absolute precursor charge. If you are analysing negative ion data, and the setting is 2, a peptide match with a charge of [M-3H]3- will be used for quantitation, while a match with a charge of [M-H]- will not.
Min. # peptides: The minimum number of valid peptide match ratios that are required in order to report a protein hit ratio.
Unique peptides only: When checked, ratios are only reported for peptide sequences that are unique to one protein hit, (which may contain more than one protein database entry). See the configuration help page for additional information.
Outlier removal: The available methods for testing and removing outliers are None, Automatic, Dixon’s method, Grubbs’ method, and Rosner’s method. Choosing auto means that Dixon’s method will be used if the number of values is between 4 and 25, while Rosner’s method will be used if the number of values is greater than 25. For a detailed description of each method, refer to the Statistical procedures page.
Peptide threshold: A peptide match will only be used for quantitation if it meets this criterion. The choice is Minimum Score, Maximum expect, At least identity (threshold), and At least homology (threshold). For the first two choices, an appropriate value must be entered in the adjacent edit box.
Normalisation: A drop down list with the choices None, Average ratio, Median ratio, and Summed intensities. A radio button is used to select the set of peptide matches to be used for normalisation. This can be all peptides, the peptides assigned to one or more protein accessions, or one or more specific peptide sequences. In all cases, only peptide matches that pass the quality tests are considered.
- None: No normalisation is performed
- Average ratio: A correction factor is applied such that the geometric mean of the ratios for the selected set of peptide matches is unity
- Median ratio: A correction factor is applied such that the median of the ratios for the selected set of peptide matches is unity
- Summed intensities: (Reporter protocol only) A correction factor is applied such that the sum of the intensities for a reporter ion peak over the selected set of peptides is the same for all the reporter ions
Report layout (Protein Family Summary)
When the quantitation method is either Reporter or Multiplex, there are several additions to the Protein Family Summary. On the Proteins tab, quantitation related information is presented on a yellow background.
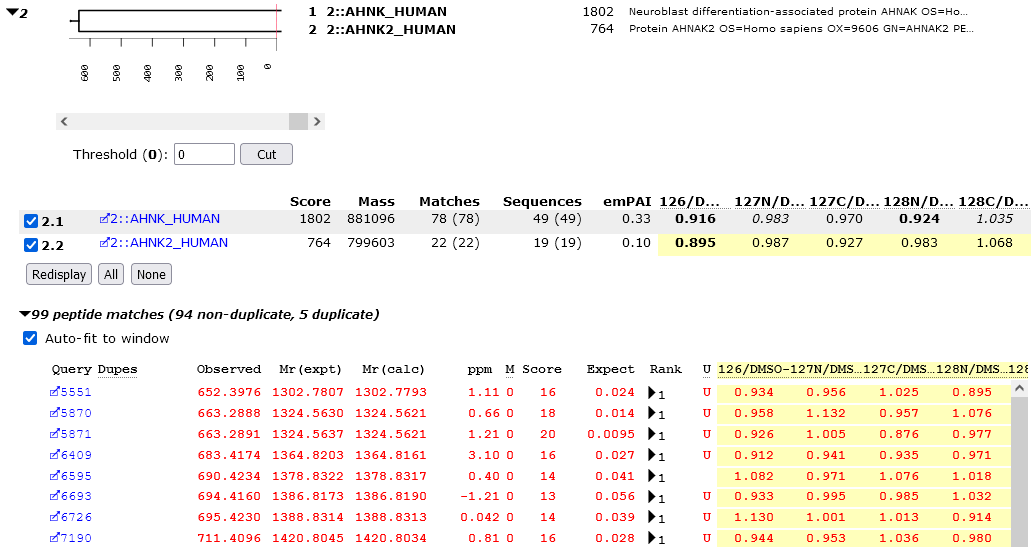
For each protein family, protein ratio(s) are displayed if the Report protein ratios checkbox is checked. Bold faced ratios are those that are significantly different from unity (p < 0.05). Other information is displayed in a tooltip (number of peptide ratios that contributed, N, the geometric standard deviation, SD(geo), and the p-value for the ratio being different from unity). If the number of valid peptide match ratios was less than the stipulated minimum, dashes will be displayed. If the peptide match ratios do not appear to be a sample from a normal distribution, the ratio will be displayed in italics, and there will be no p-value.
Since we are dealing with ratios, the average is the geometric mean and the standard deviation is the geometric standard deviation, which is a factor. In other words, the confidence interval is obtained by dividing and multiplying the average by the standard deviation, which is never less than 1.0. For example, if the average is 1.055 and SD(geo) is 1.028 then the 95% confidence interval is 1.026 to 1.084.
Warning: The standard deviation reported here measures the variance within the data submitted for a single Mascot search. This will often be tiny compared with the variance between technical replicates, (i.e. repeated analyses of this same sample). This, in turn, will invariably be tiny compared with the variance between biological replicates, (analyses of samples from different subjects or different treatment groups). It can be dangerous to read too much into small changes within a single search when the biological variance is huge. For a more detailed discussion, see Karp, N. A., et al., Impact of replicate types on proteomic expression analysis, Journal of Proteome Research 4 1867-1871 (2005).
Individual peptide match ratios will be displayed if the Report peptide ratios checkbox is checked. Dashes are displayed when a ratio cannot be determined. This may be because one or more of the relevant peaks were missing, giving a ratio which was zero, infinity, or indeterminate. Alternatively, the peptide match may have been rejected on quality grounds. For example, a disallowed charge state or modification. If a ratio is negative, which indicates some instrument or peak detection problem, (or an inappropriate correction), this is reported. However, a negative ratio is discarded when calculating the protein ratio.
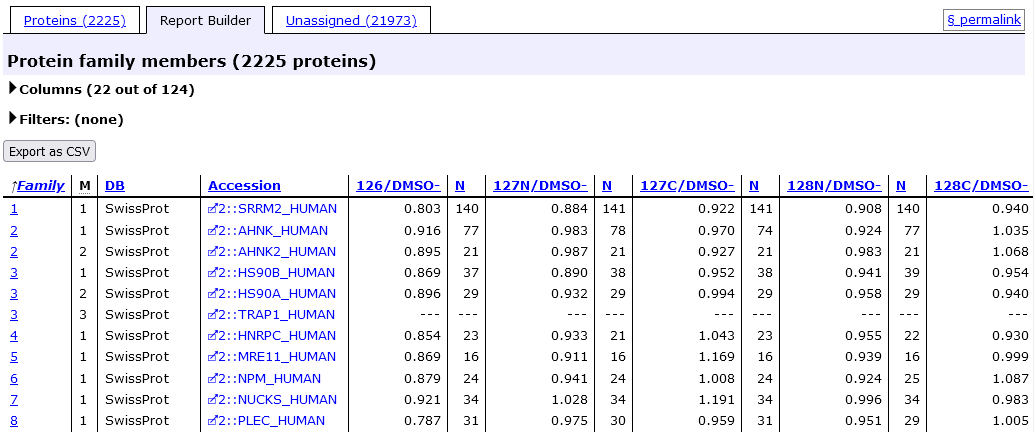
Protein quantitation information is also available on the Report Bulder tab. Expand Columns to select which values should be tabulated.
URL Switches
There are a number of switches to control the presentation of quantitation results. The default values are set in the quantitation method and can be changed in an individual report using the format controls, or by appending the relevant switch to the report URL. Switches take the form label=value and the delimiter between switches is an ampersand (&). For example, if the report URL was:
http://local-server/mascot/cgi/master_results_2.pl?file=../data/20240121/F001847.msr
The protein ratio type could be changed by appending "_quant_protein_ratio_type=average":
http://local-server/mascot/cgi/master_results_2.pl?file=../data/20240121/F001847.msr&_quant_protein_ratio_type=average
Labels and values are not case sensitive. Note that many labels begin with an underscore character. Values that are not literal strings are shown in italics.
| URL | Value | Description |
|---|---|---|
| _quant_protein_ratio_type | average | The protein ratio is the geometric mean of the peptide ratios |
| median | The median peptide ratio is selected to represent the protein ratio | |
| weighted | For each component, the intensity values of the assigned peptides are summed and the protein ratio(s) calculated from the summed values. | |
| _quant_norm_method | none | No normalisation is performed |
| average | For each peptide ratio, a correction factor is applied such that the geometric mean of the ratios for all peptide matches that pass the quality tests is unity | |
| median | For each peptide ratio, a correction factor is applied such that the median of the ratios for all peptide matches that pass the quality tests is unity | |
| sum | A correction factor is applied such that the sum of the intensities for a reporter ion peak over all peptide matches that pass the quality tests is the same for all the reporter ions | |
| _quant_report_detail | 1 | Set to 1 to display ratios for individual peptides |
| _quant_outliers_method | none | No outlier removal |
| auto | Use Dixon’s method if the number of values is between 4 and 25, Rosners if 26 or more | |
| dixons | Outlier removal using Dixons method | |
| grubbs | Outlier removal using Grubbs method | |
| rosners | Outlier removal using Rosners method | |
| _quant_min_num_peptides | N | The minimum number of valid peptide match ratios that are required in order to report a protein hit ratio |
| _min_precursor_charge | N | A peptide ratio will not be reported if the precursor charge is less than this value |
| _quant_pep_threshold_type | minimum score | Threshold for calculating a peptide ratio is a minimum score (value specified in _quant_pep_threshold_value) |
| maximum expect | Threshold for calculating a peptide ratio is a maximum expect (value specified in _quant_pep_threshold_value) | |
| at least identity | Threshold for calculating a peptide ratio is at least identity | |
| at least homology | Threshold for calculating a peptide ratio is at least homology | |
| _quant_pep_threshold_value | N | Value for threshold type specified in _quant_pep_threshold_type |
| _quant_unique_pepseq | 1 | If set to 1, ratios are only reported for peptide sequences that are unique to one protein hit, |

