Finding unsuspected modifications in narrow-window DIA data using the Mascot Error Tolerant Search
Mascot’s spectrum-centric approach doesn’t have the constraints on variable modifications, missed cleavages and protein sequences that are required by peptide-centric DIA tools. In fact, it’s nearly trivial to identify unsuspected variable modifications in narrow-window DIA data using the Mascot Error Tolerant Search.
There can be many reasons for failing to get a significant sequence match to an MS/MS spectrum. Three common reasons are:
- Enzyme non-specificity
- Unsuspected chemical & post-translational modifications
- Peptide sequence not in the database
The Error Tolerant search is a two-stage process. A standard, first pass search is performed using the search parameters specified in the form. From the results of the first pass search, all of the database entries that contain one or more significant peptide matches are selected for an error tolerant, second pass search. At the completion of the second pass search, a single report is generated, combining the results from both passes. There are some constraints on the first pass search, which are described in the help.
We took one of the prostate cancer patient files (P1.raw) from the PRIDE dataset used in the previous blog article and carried out an error-tolerant search. The spectra were acquired in DIA mode with 8 m/z isolation window. The search settings used for the first pass were the same as used before. Mascot’s statistical model for error-tolerant matches helps with assessing match quality and thresholding. A target PSM FDR of 1% was selected – this was applied to both the first and error-tolerant search passes. Significant PSM and sequence counts for the first-pass and combined search results are shown in table 1 below:
| No. significant PSMs | No. significant sequences | |
|---|---|---|
| First pass | 37231 | 5829 |
| Combined first pass + ET | 44066 | 6901 |
As you can see, the error tolerant search has found an additional 6835 significant PSMs and an addition 1072 peptide sequences at a 1% PSM FDR. The Mascot report includes a break-down of the modification statistics across the reported protein families, including identified error-tolerant matches. Let’s take a closer look at some of the possible matches.
Non-specific cleavage
The top error-tolerant match reported for this dataset is “Non-specific cleavage” with 1908 total matches. During the error tolerant search pass, the selected enzyme becomes semi-specific, (that is, only one end of a peptide needs to match the cleavage specificity), and the value of the missed cleavage parameter is increased by 1. Figure 1 below shows some examples from protein family 2 in the report (Immunoglobulin kappa constant).
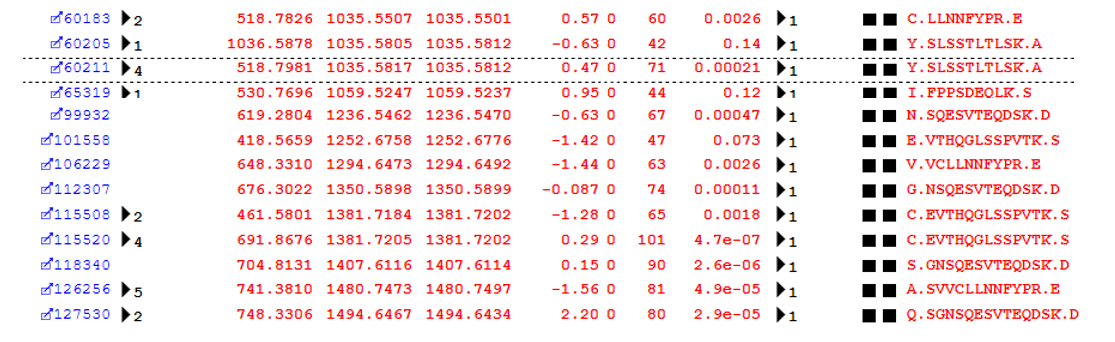 Figure 1: Examples of semi-specific cleavage peptides identified by the error-tolerant search pass
Figure 1: Examples of semi-specific cleavage peptides identified by the error-tolerant search pass
An error tolerant search is a highly efficient way to pick up these non-specific cleavage products, which would be missed by a spectral library based search which did not include the semi-specific peptides.
Unsuspected chemical modifications
The list of modifications identified in the error-tolerant search includes a large number of possible chemical modifications. Many of these are believable – for example, there are 104 additional PSMs identified with Carbamidomethylation on residues other than C. This sample was alkylated with iodoacetamide and the matches represent known artefacts of over-alkylation, so the assignment is very believable. Others assignments give a convincing sequence match but the explanation for the mass delta may require a little more thought – you can find an example of these types of matches on our error tolerant search help page.
This is Deamidation, my plus-one
Looking at the table of modifications, we can see a lot of possible error-tolerant assignments of approximately +1Da, shown in figure 2 below.
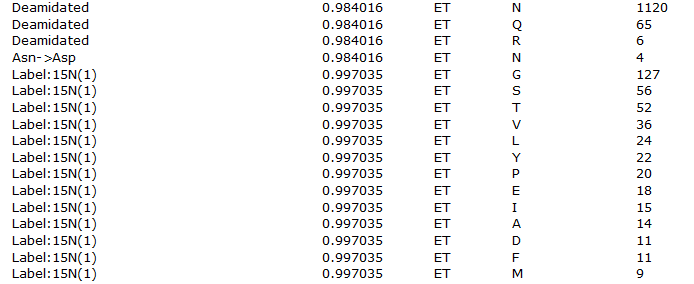 Figure 2: List of error tolerant modifications identified with a mass of approximately 1Da
Figure 2: List of error tolerant modifications identified with a mass of approximately 1Da
We discussed the issue in "The plus one dilemma", and the same considerations highlighted there apply here. Deamidation is, of course, a fairly believable assignment, but the Label:15N(1) assignments are not as this is an unlabelled sample – these are much more likely to be one of the other possibilities such as deamidation or perhaps an incorrectly picked 13C precursor.
Post translational modifications
Unless you’ve carried out an enrichment for a specific PTM, such as enriching for phosphopeptides, the error-tolerant search can be the most efficient way to identify less common post-translational modifications of interest in your sample. The error tolerant search of this dataset has identified a number of possible PTMs of interest:
Phosphorylation
One biologically important post-translational modification is Phosphorylation. The error tolerant search pass identified 126 possible phosphopeptide PSMs. As an example, we’ll take a look at an identified phosphorylation at Serine 234 in Osteopontin. As figure 3 below illustrates, we have multiple matches to the phospho-peptide, which is exactly what we’d expect to see from the data-independent acquisition of an abundant peptide – a number of matches across the entire elution peak:
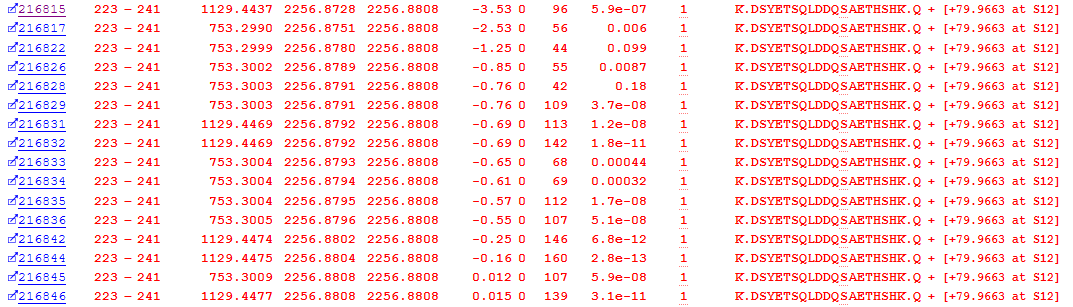 Figure 3: Matches to DSYETSQLDDQSAETHSHK with an identified phospho-serine (S12 in the peptide is S234 in the protein)
Figure 3: Matches to DSYETSQLDDQSAETHSHK with an identified phospho-serine (S12 in the peptide is S234 in the protein)
Looking at an example match, we can see that these are strong matches, and, from the Mascot Delta Score site analysis, there’s very little ambiguity regarding the site of phosphorylation:
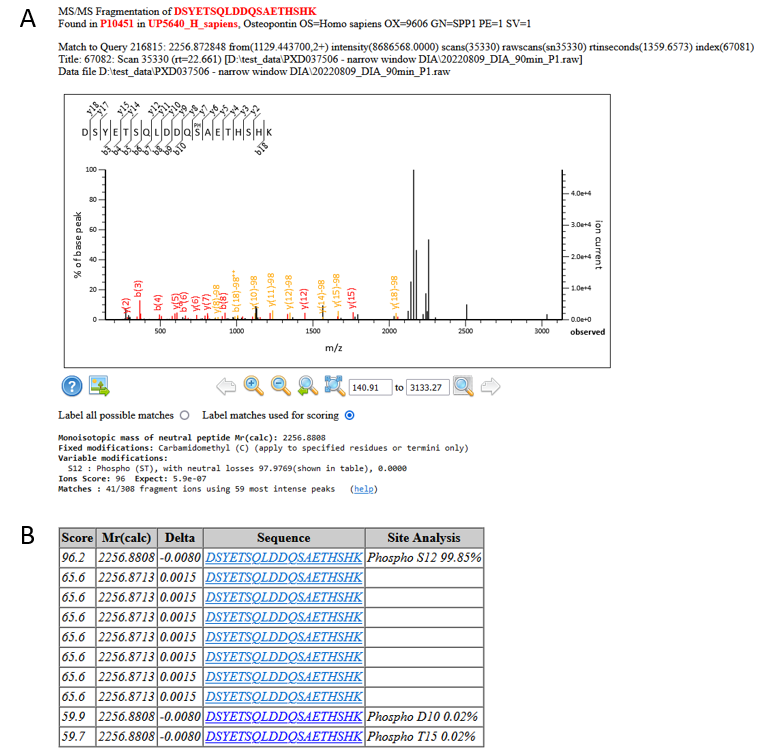 Figure 4: A) An example match and site localisation for DSYETSQLDDQSAETHSHK with S12 phosphorylated. B) Site localisation table using the Mascot Delta Score
Figure 4: A) An example match and site localisation for DSYETSQLDDQSAETHSHK with S12 phosphorylated. B) Site localisation table using the Mascot Delta Score
If we take a look in Uniprot, S234 is a know phosphorylation site on Osteopontin, so we can feel confident about the matches.
Glycosylation
Another biologically important post-translational modification is glycosylation. Mascot uses Unimod as the modifications database, which contains a number of glycans, and these are included in the error-tolerant search. The list of modifications contains a 383 possible glycopeptide PSMs. Figure 5 below shows an example from Alpha-2-HS-glycoprotein, with Hex(1)HexNAc(1)NeuAc(1) at Serine 346:
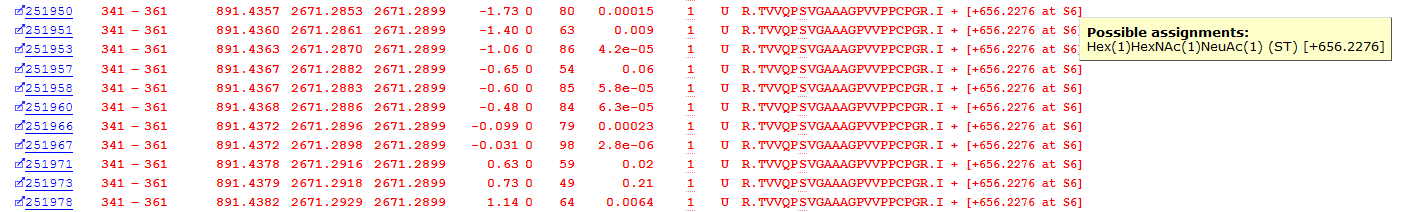 Figure 5: Error tolerant matches to a glycopeptide from Alpha-2-HS-glycoprotein
Figure 5: Error tolerant matches to a glycopeptide from Alpha-2-HS-glycoprotein
As with the example phosphorylation above, we have multiple sequential matches to the glycosylated peptide. Site localisation is, however, trickier because the matches in the spectra are mainly to the neutral loss of the glycan, meaning that we can’t determine the site of the glycan using the Mascot Delta Score – it could be either S346 or T341.
 Figure 6: A) An example match and site localisation for TVVQPSVGAAAGPVVPPCPGR with Hex(1)HexNAc(1)NeuAc(1). B) Site localisation table using the Mascot Delta Score
Figure 6: A) An example match and site localisation for TVVQPSVGAAAGPVVPPCPGR with Hex(1)HexNAc(1)NeuAc(1). B) Site localisation table using the Mascot Delta Score
Uniprot is less helpful this time, as both T341 and S346 are listed as possible O-linked glycosylation sites:
 Figure 7: Glycosylation site information from Uniprot for Alpha-2-HS-glycoprotein
Figure 7: Glycosylation site information from Uniprot for Alpha-2-HS-glycoprotein
GlyConnect confirms the assignment of Hex(1)HexNAc(1)NeuAc(1) at S346 as experimentally verified, so if you were pushed for an answer you might elect for S346, but from the MS/MS matches from Mascot we can’t definitively say one way or the other as to the actual site.
Primary sequence variants
The final class of unsuspected variation is single residue substitutions in the primary sequence of a peptide. The error-tolerant search of this dataset found 922 possible PSMs with single residue substitutions. An example of this, a Proline to Serine substitution at residue 182 in Immunoglobulin heavy constant alpha 2, is shown in Figure 8 below:
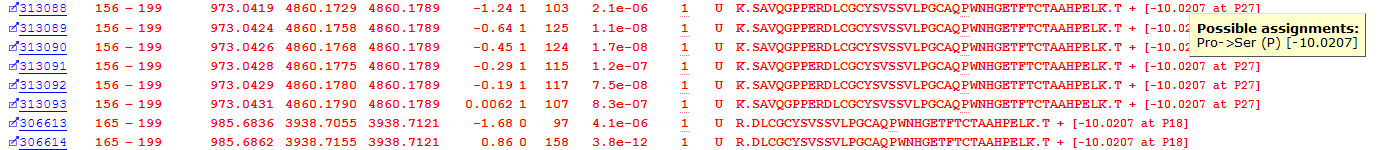 Figure 8: Matches to a primary sequence variant at P182 in Immunoglobulin heavy constant alpha 2
Figure 8: Matches to a primary sequence variant at P182 in Immunoglobulin heavy constant alpha 2
We have multiple, high scoring matches to the same residue substitution, and the site localisation using the Mascot Delta Score is strong:
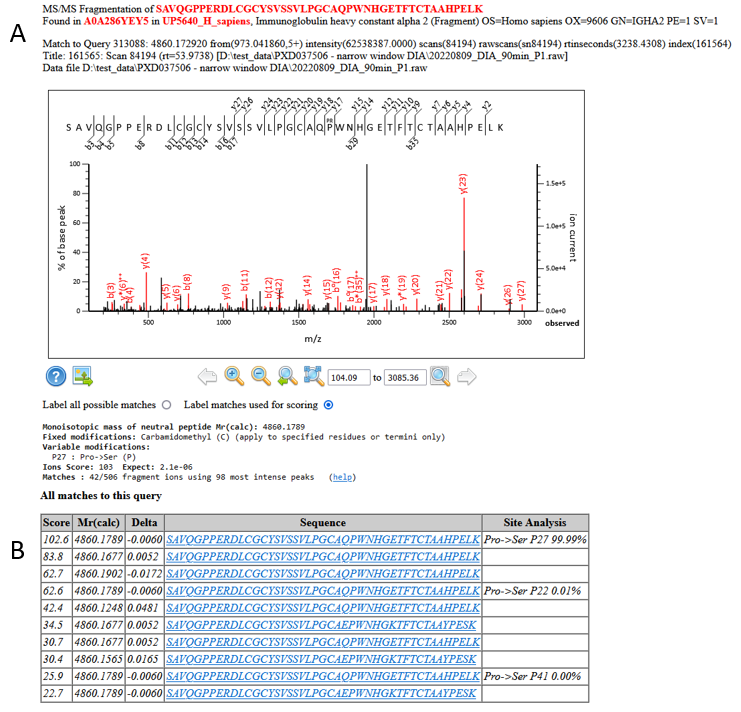 Figure 9: A) An example match and site localisation for the residue substitution Pro->Ser at P182. B) Site localisation table using the Mascot Delta Score
Figure 9: A) An example match and site localisation for the residue substitution Pro->Ser at P182. B) Site localisation table using the Mascot Delta Score
The EBI’s ProtVar lists Proline to Serine at residue 182 as a known variant, so we can be very confident in this assignment.
Conclusions
Including all the possible variants covered by the error-tolerant search in a spectral library based search would be impractical, whether the library was experimentally derived or generated in-silico. Running an error tolerant-search on the single DIA file used here demonstrates that this approach is potentially a powerful way to identify unsuspected sequence variants, modifications and non-specific enzyme cleavage products in a chimeric narrow-window DIA datasets as well as DDA.
Keywords: chimeric spectra, DIA, error tolerant, variable modifications
Hello, in this blog post I can read “it could be either S346 or T341″ and the peptide in figure et_dia_blog_figure5.png is fully tryptic: but would trypsin cut at R340 if the T341 would be glycosylated?
Thanks
There’s likely to be steric hindrance, which would suggest S346 as the glycosylation site. There’s no evidence either way in the actual fragmentation data though, which is what the text is referring to.