Ion series for EThcD
The ion series considered during a Mascot search are selected by choosing an instrument type. If you have Mascot Server in-house, you can edit existing instrument types and create new ones. There are 17 ion series available, and Mascot takes an iterative approach to scoring. If the number of matched peaks in a particular ion series is no better than would be expected by chance, the series is dropped before the final score is calculated.
This might lead you to think it best to include all of the series, all of the time, rather than figuring out which ones matter. This is actually not a good idea because selecting all series increases the number of degrees of freedom – the scope for getting chance matches – without any benefit for the real matches. Including unnecessary series causes the score distribution for false matches to broaden and shift upwards, while that for the true matches stays the same, so that discrimination is reduced and the sensitivity at a given FDR is reduced. Better to be selective, which is why we have multiple instrument types.
Inspecting a few strong matches to decide whether there is evidence for a particular series can be misleading because different peptides within a single data set can behave very differently. The safest way to decide whether a particular ions series should be added or dropped is to look at how this affects sensitivity on a large and representative dataset. To illustrate, let’s look at some EThcD data. Electron-Transfer/Higher-Energy Collision Dissociation was introduced in 2012 by Albert Heck’s group. The combination of b/y and c/z fragment ions in a single spectrum can assist in identification by database searching and often improves site localisation for modifications.
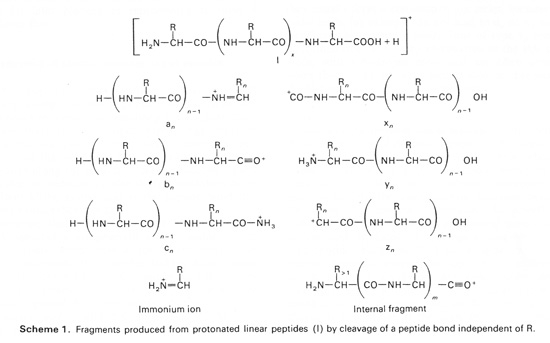
Although some publications suggest that special software is needed to search EThcD data, this is not correct. The c and z series have the same structures as those found in ECD data, and have been available in Mascot since version 2.2, more than 10 years ago. Possibly, there is some confusion associated with ion series nomenclature. We adhere to the nomenclature established many years ago by Biemann’s group, as can be seen by comparison of the figures on the Mascot help page for MS/MS fragmentation with the figure below, which is Scheme 1 from Biemann’s 1988 review paper "Contributions of mass spectrometry to peptide and protein structure". Similar figures in many other papers from his group contain identical information. Note that the Biemann notation is a departure from the earlier Roepstorff and Fohlman notation, which used capital letters. The composition of a (Biemann) y ion is that of a (Roepstorff) Y+2 or Y” ion.
We downloaded a set of EThcD raw data files from the Heck lab. paper "Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry". This set corresponds to the analysis of lysed HeLa cells digested with trypsin. The raw files were peak picked using Mascot Distiller and searched using these settings: Trypsin/P with 2 missed cleavages, Carbamidomethyl (C) fixed, Oxidation (M) variable, Peptide tolerance 15 ppm, fragment tolerance 30 ppm.
Different combinations of ions series were tried to see which gave the best sensitivity at 1% FDR. To some extent, the selections were guided by experience – it would not be feasible to make an exhaustive analysis of all 2^17 possible combinations. The difference in sensitivity between the optimum set of ion series and an instrument containing all ion series is actually quite small.
| Instrument | Ion series | PSM count 15ppm/30ppm | PSM count 0.5Da/0.5Da |
|---|---|---|---|
| All | a a* a0 b b* b0 c x y y* y0 z z+1 z+2 d v w | 58489 | 46814 |
| EThcD | a a* a0 b b* b0 c y y* y0 z+1 z+2 w | 59221 | 49642 |
The difference in PSM count at 1% FDR is only some 1%. Mainly because this is high accuracy data being searched with narrow mass tolerances, so the presence of unnecessary ion series doesn’t lead to large numbers of false fragment ion matches. If we search with wider mass tolerances, 0.5 Da for both peptide and fragments, the effect becomes stronger, with a difference in PSM count at 1% FDR of nearer 6%.
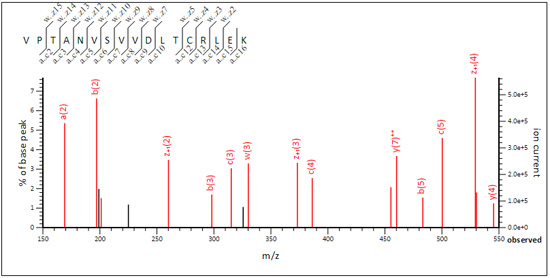
The optimum instrument type still contains an unusually broad range of ion series. This is because some EThcD spectra tend to be ETD-like and others are more CID-like. The results are here if you want to take a look. For example, in family 1 (G3P_HUMAN), VPTANVSVVDLTCRLEK (query 78391) is ETD-like.
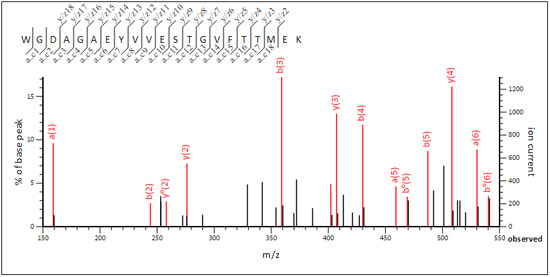
While WGDAGAEYVVESTGVFTTMEK (query 89737) is CID-like.
In peptide view, choose label all possible matches to get a more complete picture of which ion series are highly populated.
If this topic is of interest, you may want to look at an earlier post concerning data sets which contain both CID and ETD scans.
Keywords: EThcD, fragmentation, ion series